
Editor’s Note: This article is being simultaneously published on our affiliate site, Mad in the UK.
In my previous article, I looked at some evidence on ‘disappointing’ long-term outcomes for early intervention in psychosis (EIP) recipients, and also some findings on ‘functional outcomes’ between drug discontinuation and maintenance treatment cohorts. I attributed these trends chiefly to the inefficacy of neuroleptics over the long-term.
I hope my discussion can contribute a little to advancing understanding of how a drug based ‘disease model’ of mental distress can often do more harm than good. The growing awareness of which owes much to Robert Whitaker’s ground-breaking work in Anatomy of an Epidemic. For a summary of the book’s arguments read this.
Here, I start by looking at some objections to ‘deprescribing’. I consider them as revealing the incoherence of certain assumptions behind early intervention, and indeed, of psychiatric diagnosis more widely.

When responding to the case that patients seem over the longer term, to have better social and occupational (functional) outcomes off standard maintenance treatment (MT), many psychiatrists will object that the risks of relapse after drug discontinuation are too great. Indeed, the dangers of withdrawal should not be taken lightly. ‘Relapses’ can have disruptive and serious consequences.
Nevertheless, most of the objections do not take into account the drug withdrawal induced ‘rebound’ effects I discussed in the previous article. The real nature of such symptoms is generally overlooked (and misdiagnosed), along with their potential avoidability. Obviously, it is imperative to minimise risks of relapse, but not at the cost of the reduced life expectancy associated with long-term use of neuroleptics.
There is a clear need for services and patients to work with more appropriate tapering regimens to enable those with remitted psychosis to reduce the drugs’ toll on their systems. Employing more gradual (‘hyperbolic’) tapers, the risks of withdrawal can be minimised and managed. The guidance which follows from this understanding can actually be considered more cautious than current standard clinical practice. For instance, when switching drugs, as well as in the protocols of pharma sponsored trials on antipsychotic efficacy. Where often four weeks or less are scheduled for drug discontinuation.
The idea of gradual tapering—with progressively smaller dose reduction increments over an extended taper period, giving the brain time to adapt—was understood informally by some in the mental health recovery community, notably Will Hall, even if it wasn’t generally recognised by prescribers, much less the majority of those who had to live with the drugs.
This kind of approach has now been given a more formal, scientific underpinning based on the ‘hyperbolic’ relation between dose level and dopamine receptor occupancy.
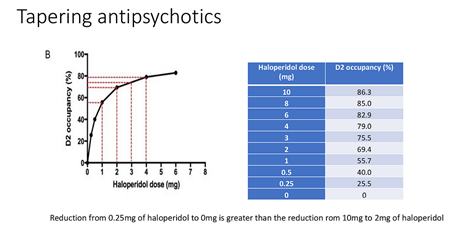
Source: Mark Horowitz PowerPoint presentation. More details here.
Still, however, ‘rebound’ symptoms from withdrawal are usually interpreted as demonstrating an ‘underlying illness’. Typically for those defending the status quo, that psychosis patients often ‘relapse’ following discontinuation shows they are subject to “vicious brain diseases”. This point seems to be more an article of faith for these psychiatrists rather than something of which they can generally give any determinate account.
Leaving drug withdrawal effects aside, some of the objections put forward against deprescribing can, I think, be rather revealing. I pick out a response by Ann Mortimer in her 2018 critical commentary in response to guidance encouraging more openness towards deprescribing antipsychotics.
While wrestling with the real issues services face, she tries to dispel findings, such as Wunderink 2013, that patients subsequently do better off meds:
In my own [EIP] service, approaching half of new patients have persistent substance-induced psychotic disorder. Although patients may later attract a schizophrenia-spectrum diagnosis, there is a flaw in any contention that such patients really had schizophrenia all the time. Early chronic substance misuse in vulnerable individuals may cause subtle brain damage that is not recoverable, even after several years of anti-psychotic treatment. This is not the same thing as having schizophrenia in the first place. Clinically, such patients usually have suffered significant adversity, which perhaps explains why they develop psychosis when, fortunately, the vast majority of young substance misusers survive without it.
Such wide ‘variability’ is a challenge that EIP services have to take on board. As Mortimer notes elsewhere, referrals to EIP services greatly outnumber what would be in-line with estimates of schizophrenia’s prevalence in the population. Evaluations of EIP services have highlighted high rates of non-cases (‘bycatch’) and the potential for misdiagnosis.
Before moving on let’s remind ourselves of the rationale for EIP programmes. Two interrelated notions were that duration of untreated psychosis (DUP) was critical in determining long term course, and that there was a ‘critical period’ of 3–5 years around typical onset of first episode psychosis (FEP), with the greatest opportunity to impact on long-term course. This ‘critical period’ is usually late adolescence/early adulthood, where greater neuronal and ‘psychosocial plasticity’ is thought to be displayed.
A related innovation was the hypothetical construct of ‘ultra high risk’ of psychosis. This sought to identify those who showed ‘prodromal’ signs of ‘attenuated psychosis’, based on symptom ratings scales. However, according to a critique of this concept by Jim Van Os et al, distinguishing such a group from those already presenting to services with anxiety, depression and other ‘milder’ mental health difficulties, often proved unfeasibly difficult.
But back to Ann Mortimer’s argument above: Firstly, she writes that not every psychosis is schizophrenia and there is a flaw in the assumption that a proportion of patients had ‘schizophrenia’ in the first place. Thereby acknowledging, that misdiagnosis of ‘schizophrenia’ is not uncommon—as indeed is the conflation of ‘psychosis’ with ‘schizophrenia’. Her position seems to be that patients who go on to do well after drug discontinuation, probably didn’t really have ‘schizophrenia’ to begin with.
On the other hand, according to her line of thought, poor outcomes in maintenance treatment cohorts indicate greater disease severity, which she takes as incompatible with being able to discontinue (or reduce). Poor outcomes in this group are due to the patients’ ‘schizophrenia’ effectively preventing drug discontinuation. Attempting to gloss over the trends in this way, reveals the term ‘schizophrenia’ as, effectively, just a shorthand for poor outcomes after first episode psychosis; a shorthand that appears unexplanatory and even self-defeating.
A conceptual dead-end can be seen to arise for EIP services and research. If a referral fares well, then potentially, it was a non-case. A ‘watch and wait’ strategy would have sufficed. On the other hand, if the patients fares poorly after early intervention, then it can be put down to their having ‘severe schizophrenia’. So there wasn’t much services could have done to prevent a disappointing outcome anyway. In short, the interventions appear either unnecessary or ineffective… So what are they preventing?
To be clear I am not suggesting that all the interventions made in EIP services are unhelpful. Providing appropriate psychosocial support and practical psychological resources certainly has value. But when one considers both low rates of ‘transition’ to psychosis in those supposedly at ‘ultra high risk’ and the high rates of non-cases that evaluations raise, reviews of EIP seem to be presenting arguments for its redundancy.
If the late adolescence/early adulthood period is genuinely ‘critical’ and ‘sensitive’ for still developing brains, then it is precisely this which should make clinicians wary of biochemical interventions!
But, it is at this stage of life that mainstream psychiatrists are most keen for intervening with psychotropic drugs; interventions that inevitably produce unintended consequences. Some are foreseeable—neuroleptics are known to alter neuronal development—but also many that will be poorly understood.
The ‘critical period’ in development is probably a contributory factor in adverse reactions to selective serotonin re-uptake inhibitors (SSRIs) in the under-25s. Higher risks in the age group were eventually recognised with black box warnings for the drugs—even though regulators overlooked the data for more than a decade before responding.*

(Source)
Ann Mortimer speculates that many referrals to EIP services, ones who later attract a schizophreniform diagnosis, could be due to subtle brain damage from early chronic drug use – “that is not recoverable, even after several years of anti-psychotic treatment”—she adds, incomprehensibly. If anything will cause brain damage, it is years of exposure to neuroleptic drugs, representing a sustained intake of neurotoxins known to cause reductions in cortical volume. Is it any wonder?
But to address the issue of street drug use: there has been much media commentary about an association between cannabis use and risk of psychosis, especially ‘skunk’ strains with higher levels of the psychoactive THC. But, such drug use and its consequences do not take place in a vacuum. Many other factors will be relevant. Moreover, plenty of FEP cases may not have had chronic drug habits before presenting, nor necessarily have experienced particularly adverse upbringings.
As I’ve mentioned, any supposed ‘high risk’ group for psychosis would tend to overlap with those who may already have presented to services with ‘milder’ mental health issues. A relevant risk factor for a psychotic break can even be consumption of psychotropic drugs like SSRIs. I’ve previously discussed how these appear to raise the risks of developing psychotic or manic symptoms.
It is acknowledged that “psychoactive drugs that do not directly impact on the dopamine system can impact on dopamine release through indirect effects”. One neuroscientific theory posits serotonergic over-expression as leading to the ‘dopaminergic instability’ theoretically associated with psychosis.
Incredibly, a recent EIP research trial looking at ‘staged treatment’ to prevent psychosis in young individuals deemed at ‘ultra high risk’ tested the SSRI fluoxetine (Prozac) as an intervention. The Prozac group actually showed slightly lower symptom rating improvements and marginally more ‘transition to psychosis’ than the placebo group. The Prozac arm had the highest reported rate for psychotic events in the trial.**
It is evident that intake of an SSRI can heighten the danger of any illicit drug use resulting in mania or psychosis. The risks of antidepressant and cannabis use can be mutually reinforcing. Studies suggest that cannabis effectively increases the concentrations of SSRIs in the body, raising their activation potential, whilst it is thought that cannabis’ psychoactive THC also acts on serotonin receptors.
Note that effects like these would constitute causal pathways to psychosis, chiefly through compounding drug effects, and lead to symptoms that would likely be assessed as ‘schizophreniform’.
A subsequent period of treatment with neuroleptics would, over time, tend to result in dopaminergic supersensitivity. So if, for example, after months or years of treatment, someone experimented with drugs like cocaine, amphetamines, or MDMA—which act as dopamine agonists—any drug induced state would be more likely to develop into a persistent psychosis. With the person being increasingly liable to being assessed as “having” some life-long psychiatric condition. This is likely to entail prescription of psychiatric drugs on an indefinite basis.
As I hope I’ve just outlined, this would be a pathway by which they “attract a schizophreniform diagnosis” due to primarily exogenous causes. None of it would imply the presence of some underlying disease.
I wouldn’t deny that in such cases there may be some level of pre-existing susceptibility; meaning that when stressors arise, some threshold (vaguely understood) is exceeded or overwhelmed. But clearly, as Ann Mortimer might say, this is not the same as “having schizophrenia” in the first place. Case histories like this with, perhaps, some susceptibility and drug—or drug withdrawal—induced changes converging, do not equate to “vicious brain diseases”.
Clearly, there is a range of other factors besides drug effects that one might point to in explaining any particular case. Psychosis is widely understood as being on a continuum with everyday experience, with much of the population (8–30%) reported as experiencing transient ‘psychotic’ experiences at some point in their lives. For example, it is acknowledged that excess sleep deprivation will initiate symptoms of a ‘psychotic’ character.
At the same time, experiences considered clinically abnormal such as ‘hearing voices’ can be lived with, in the course of productive and worthwhile lives. Such ‘symptoms’ may be managed and integrated without significant distress or dysfunction. As Jim Van Os concluded, ‘attenuated psychotic symptoms’ are commonplace. The rates of ‘transition’ to clinical psychosis being much as they are for the population in general.
Given that assessments of ‘psychosis’ are made on the basis of behavioural responses, and from the perspectives and interpretations of clinicians, I suggest that psychosis should be understood as a functionally and indeterminately defined state—potentially realized in the central nervous system by multiple causal mechanisms.
The quest for an explanation of ‘psychosis’ and ‘schizophrenia’ in terms of single variables has proved unsuccessful and harmful. Even reductive neuroscientific approaches now posit serotonin and glutamate signalling as playing roles ‘upstream’ of instability in dopamine pathways. Furthermore, some recent neuroscientific findings indicate that much ‘treatment resistant’ first episode psychosis cannot be associated with ‘elevated levels of presynaptic dopamine synthesis’—the dopamine theory’s more recent iteration.
The history of attempts at biochemical reduction have eventually led to a scientific interpretation of psychosis as “a network dysfunction that includes a variety of brain regions (and multiple neurotransmitter pathways), of which impairment at any level could precipitate symptoms.”
All this would rather suggest, not some discrete illness or disease process, but perhaps some overlap of susceptibilities to such anomalous functioning; including, for example, higher levels of risk arising from prenatal or perinatal complications, winter birth, or experience of childhood abuse, etc. Their joint presence amounting to a cluster of predispositions, potentially triggered or exacerbated by a wide range of external stressors; social adversity, environmental insults, urbanicity, etc. As these later risk factors present stronger correlations than any genetic associations that have been pointed to, this would underline the inappropriateness of any assumption of an underlying ‘brain disease’.
The failure of the ‘high risk’ concept and wide variability of course and outcome suggest that the ‘schizo’ paradigm—inasmuch as it assumes some reducible disease process, separate and distinct from other dimensions of functioning and well-being—can be safely dispensed with. And moreover, that it is has become an obstacle to clearer understanding. This is not to mention both the social stigma and the disheartening effect diagnosis may have on a person’s aspirations in life.
Although there has been some signs of a growing awareness that ‘schizophrenia’ is probably not a scientifically valid concept, it does seem that the old assumptions are still driving routine clinical practice. These widespread and erroneous assumptions may stem, in part, from decades of very influential drug company marketing, based on flawed research trials and publication bias.
Notes
* Critics like Peter C. Gøtzsche are, no doubt, right to point out that these risks of SSRIs extend to all age groups.
** The trial’s researchers, among them Patrick McGorry, who had pioneered the development of the ‘early intervention’ model, believed the poor results were likely due to “real-world problems of treatment fidelity and adherence”. But it is astounding to me that the researchers believed Prozac could prevent psychosis. Such a flawed choice entails, effectively, in my view, that the trial doesn’t provide good evidence, either for or against its hypothesis. Although it does further support the assessment that SSRIs raise risks of mania and psychosis.











Thank-you, Neil. THANK-YOU…. Oh, God, if only I and THEY knew THEN, what we are learning NOW, maybe a whole lot of suffering could have been avoided….
Report comment
Thanks. I know the feeling.
Report comment
I’m not certain how many times I must point out that the psych drugs can create the “invalid” psych DSM “bible” disorders.
https://psychrights.org/2013/130429NIMHTransformingDiagnosis.htm
I agree, Robert Whitaker did an excellent job of pointing out that the antidepressants and ADHD drugs can create the “bipolar” symptoms, in ‘Anatomy of an Epidemic.’ We donate to MiA almost every year, in thanks for Robert’s research findings, and his honest reporting.
But I’ll repeat, once again, my research findings … which are relevant, Neil, to your concerns of “schizo” diagnoses, and “psychosis,” in general.
The antipsychotics / neuroleptics (as well as the antidepressants, since they’re also an anticholinergic drug) can create the positive symptoms of “schizophrenia” (including “psychosis”), via anticholinergic toxidrome.
https://en.wikipedia.org/wiki/Toxidrome
Plus, the neuroleptics can also create the negative symptoms of “schizophrenia,” via neuroleptic induced deficit disorder.
https://en.wikipedia.org/wiki/Neuroleptic-induced_deficit_syndrome
The psych DSM “disorders” are iatrogenic illnesses, created with the psych neurotoxins … not “genetic disorders.”
Report comment
The Mortimer article has this statement at the end;
“DECLARATION OF INTEREST
A.M. has worked with pharmaceutical companies regarding the introduction of atypical antipsychotic treatments for schizophrenia, received research funding and spoken at conferences.”
Tells me all I need to know. The arguments against deprescribing and in favour of drug based early intervention services read like drug company marketing and face saving arguments dressed up as science.
Report comment
Have you researched psychosis in bi-polar individuals? I’m not an expert in the DSM system, but it seems odd to me, that psychosis can present in both schizophrenia and mania.
In critiquing treatment choices, it would also seem important to research pressure from patients and their families to banish the psychosis symptoms, and other symptoms be damned.
Report comment
I havent looked much at diagnostics. I looked a little here at the overlap. https://www.madintheuk.com/2023/04/effect-ssris-adolescents/
Report comment
“If the late adolescence/early adulthood period is genuinely ‘critical’ and ‘sensitive’ for still developing brains, then it is precisely this which should make clinicians wary of biomedical interventions!”
This kind of critical thinking (not to mention common sense) is woefully absent in psychiatry. I don’t know why the obvious never occurs to most of them. Wait a minute! Yes I do! Their confirmation bias has them primed to see what they’re looking for, even if they’re not sure what it is they’re looking at, i.e. ‘schizophrenia’, or ‘depression’, or ‘bipolar’, or ‘anxiety disorder’, or any other ‘disorder’ they happen to think of…
Therefore, the last thing any medical doctor should do is reach for their prescription pad.
Report comment
Thanks!
Report comment
Thank you! 🙂 I enjoy reading your articles very much.
Report comment
Neil,
Let me tell you how SSRIs cause harm. If you look at the NIH page on medications that cause nutrient deficiencies, it lists SSRI’s and the nutrients it alters include Folate…and on the column that lists “risk factors” it lists MTHFR mutations.
Why is that important? Because double MTHFR mutations, (especially homozygous C677T) cause a reduction in your ability to metabolize Folic Acid. So if the mutation causes you to have low Folate and you take SSRIs which also affect Folate, that’s going to affect your ability to recycle homocysteine to methionine and make SAMe, which donates part of itself for multiple important processes, including manufacture of neurotransmitters, detoxification by the liver, DNA methylation and more. (This process is called 1-Carbon metabolism).
By the way, marijuana lowers Folate. So if you have low folate caused by MTHFR mutations and add daily, heavy use of marijuana, you won’t have enough Folate to recycle homocysteine, which is toxic to neurons.
I visited the group Surviving Antidepressants and was shocked to see that the so called “withdrawal symptoms” are exactly the same as the symptoms caused by MTHFR mutations and high homocysteine, including atrial fibrillation, ear pain, tinnitus, neuropathy, fatigue, pain, rashes, anxiety, weakness, akathisia, heat intolerance, etc
It makes sense, when you decrease the dose of SSRIs (which affect Folate) you dysregulate 1-Carbon metabolism.
I asked them if they’d had an MTHFR test and they showed me a thread that confirmed my suspicions. All who posted their results were positive, mostly C677T homozygous, no negatives.
I’m giving you guys the answer you’ve been looking for. Let’s see how long it takes for someone to listen. Let me know if you want sources.
Report comment
Re schizophrenia….look up “schizophrenia and MTHFR”. I left some links on Peter’s “Looking for the psychiatric yeti” article.
Report comment