

Editor’s Note: In this essay, neuroscientist Peter Sterling provides an “overview of what we don’t know about the brain regarding mental disturbance and what we should not be doing to the brain physically and chemically as ‘therapy.'”
Here I comment on the 2022 review, “Causal Mapping of Human Brain Function” by Siddiqi et al., which appeared in Nature Reviews Neuroscience. I place its reasoning in the historical context of psychosurgery and other physical manipulations of brain function—and also in the present context of new understandings from brain imaging, genome-wide studies, and longitudinal studies of mental disturbance. Here are the Review’s stated goals:
This Review focuses on the objective of symptom localization, which aims to identify causal links between symptoms and neuroanatomy. When a symptom is successfully localized, it may potentially be treated by modulating the corresponding neuroanatomy. The second objective, mapping the direction of information flow, aims to understand how one brain region can causally influence another. These experiments attempt to estimate the direction of information flow between two or more nodes in the brain using various measures of effective connectivity…Similar approaches have now been used to map the causal neuroanatomy of movement disorders, mood disorders, anxiety-related disorders, psychotic disorders, disorders of consciousness and various other neuropsychiatric phenomena.
The Review opens by considering identified neurological disorders with identified causes and identified therapies grounded in identified neuroscience. Its primary exemplar is Parkinson’s disease, whose immediate cause is loss of dopaminergic neurons in the substantia nigra, and whose treatment involves providing the dopamine precursor to the remaining dopaminergic synapses so that they continue making and releasing dopamine in response to natural stimuli as part of the natural circuit. All good. But then the Review pulls a fast one: it inverts the argument.
Whereas neurology starts with known damage that causes known symptoms and finds effective therapies based on known neuroscience, this Review starts with mental disturbances that it claims to be symptoms of an underlying “brain disorder”, which it claims to “localize neuroanatomically” in order to “treat” with brain manipulations for which there is no foundation in neuroscience. Thus, the Review reasons by asserting an equivalence between something not known to something that is known, apparently hoping that the difference will go unnoticed.
To be clear: there is no neuroscience to suggest that any mental function would be improved by ablating or stimulating a particular structure in the prefrontal cortex or its associated subcortical regions. To the contrary, what we know of the intrinsic organization of the prefrontal cortex suggests that mucking with it is unlikely to help. Any suggestion to the contrary is simply wild speculation. The Review tries to justify its story by claiming efficacious results. But I have heard these claims before, and they never check out. They’re just a succession of Ponzi schemes, as here recounted.
Some history of mental disturbance and its desperate “cures”
My engagement with the problem of mental disturbance and its desperate cures started in 1972, early in my career at UPenn, when a visitor shared a piece in the NY Post about lobotomies being performed on children. Incredulous, I searched and found the published medical reports. Moreover, these were not isolated cases. Frontal lobotomy and other forms of “psychosurgery”, which I assumed had been abandoned in the 1950s, were still being performed at Penn and other Philadelphia medical schools. In 1967 and 1969 Dr. Robert Groff, Chair of Neurosurgery, performed successive prefrontal lobotomies on a young man, Stanley Chase, leaving him with a right-sided hemiplegia and a deteriorating IQ due to a communicating hydrocephalus caused by subdural bleeding.
In the early 1970s, Jewell Osterholm at Philadelphia’s Hahnemann Hospital performed anterior cingulotomy to treat drug addiction; Thomas Ballentine at Harvard’s Mass General Hospital performed cingulotomies to treat depression and chronic pain; William Scoville at Yale performed frontal lobotomy and then bilateral temporal lobectomy to treat schizophrenia; Vernon Mark at Mass General was performing amygdalotomy to treat “violent” behavior—to name just a few. I published my understanding of these practices (Sterling, 1978) and testified in Federal District Court on behalf of Stanley Chase (which was reported in the Philadelphia Evening Bulletin, November 1, 1975).
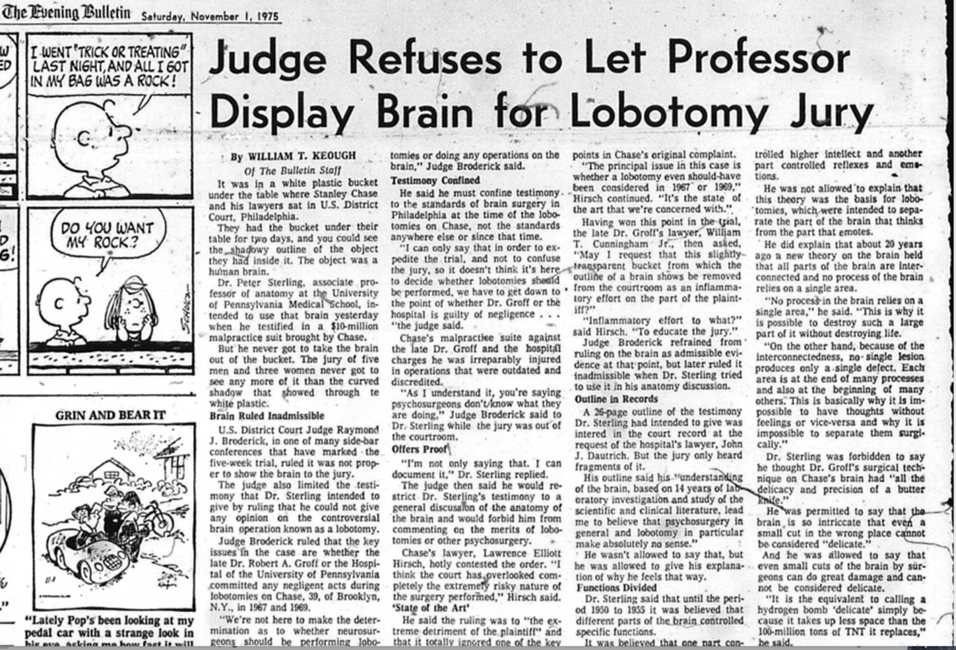
The list of symptoms treated by prefrontal lobotomy, leucotomy (cutting tracts), cingulotomy, amygdalotomy and so on, included depression, mania, obsessive-compulsive behavior, addiction, chronic pain, criminal behavior and violent behavior. The reasoning always began by noting a brain area that, when damaged by accident or experimentally in a non-human primate, calmed the subject and cheered them up. That was the “causal reasoning” from the outset—1935—forward. Surgeons reported glowing results; for one example, Geoffrey Knight, a leading British neurosurgeon, reported that one of his depressed patients, following the bilateral disconnection of her frontal lobes, could now experience “normal mother love”. Right.
Following thousands of such reports, Life magazine had printed a story in 1947, “Psychosurgery: Operation to Cure Sick Minds Turns Surgeon’s Blade into an Instrument of Mental Therapy”, and a Nobel Prize was awarded to the inventor of the lobotomy in 1949. But the claimed benefits faded with time. Then patients had to fend for themselves with what remained of their wits. In short, psychosurgery didn’t work. Moreover, it violated medicine’s key principle: first, do no harm. Because of psychosurgery’s therapeutic failure, it declined for several decades (Sterling, 1978; Valenstein, 1986; 1987).
But now all this has been forgotten, and a new generation of psychosurgeons has been emboldened. Moreover, they are supported by prominent journals that publish the same old single-case reports that claim good results, even when casual inspection indicates they are clearly not good. Consider these excerpts from a recent “case report” published by Frontiers in Neuroscience (He et al, 2022):
Somatic symptom disorder (SSD) is a form of mental illness that causes one or more distressing somatic symptoms leading to a significant disruption to everyday life, characterized by excessive thoughts, feelings, or behaviors related to these symptoms… Considering the patient’s financial status, and her inability to have frequent, long-term, face-to-face follow-ups…she and her family opted for bilateral anterior capsulotomy…
The report explains that “anterior capsulotomy” [bilateral lesion of the anterior limb of the internal capsule] is a point of convergence for key white matter fibers connecting the prefrontal and anterior cingulate cortices with the hippocampus, amygdala, and thalamus. In other words, the site is a roundabout for the most critical structures in the human frontal lobe. Note that this patient, in the People’s Republic of China, is apparently too poor to afford sustained psychotherapeutic care. The report continues:
The patient’s somatization, depression, and anxiety symptoms as well as quality of life improved significantly and steadily… However, the patient developed new somatization symptoms, including dizziness, headache, and sternal pain, 10 months after the operation. Therefore, the patient resumed taking flupentixol and melitracen in order to control the new symptoms. This study shows that bilateral anterior capsulotomy appears to be a complementary treatment for refractory SSD with depressive and anxiety symptoms… In this report, one patient with refractory SSD accompanied by depression and anxiety was effectively treated.
But how to square that conclusion with this acknowledgment?
As no preoperative personality-related assessment was conducted, it was not possible to accurately determine whether the patient’s personality changed. In addition, the patient developed new somatic symptoms 10 months after the surgery. Although the new somatic symptoms were brought back under control with medication, their long-term efficacy is worth exploring in a longer follow-up study. Interestingly, the use of flupentixol and melitracen before surgery failed, whereas it effectively controlled the symptoms after surgery. Therefore, the mechanism(s) underlying restoration of the therapeutic effects of the medication following internal capsule injury of the forelimbs need to be determined.
Why does Frontiers publish this atrocious nonsense? I believe that it represents deep corruption that is creeping back into neuroscience—as will be further noted below.
This report resembles many from the psychosurgeons of the 1970s, such as Ballentine, whose cases were reviewed by Teuber et al (1976), and other cases reviewed by Mirsky and Orzack (1976), all summarized with full references in Sterling (1978). The pattern is stereotyped: claims of short-term response with subsequent relapse and further treatment with drugs. The pattern resembles that of electroconvulsive therapy (ECT), which is widely acknowledged to cause memory loss. Practitioners of ECT deny that it causes much long-term loss of memory, and they preserve deniability by failing to perform simple memory assessments in advance—as first reported by Irving Janis in 1950, who found substantial losses (Sterling, 2000, 2001). As practitioners of ECT switch over to transcranial magnetic stimulation (TMS), they finally acknowledge that it is “much safer”. That is, they finally admit that ECT is not safe. But TMS is just now taking off, so whether it is also unsafe may not be apparent for a decade.
Now neurosurgeon Veerle Visser-Vandewalle and 41 colleagues (2022) propose deep brain stimulation (DBS; a practice which involves drilling holes in the skull and threading electrical wires deep into the brain tissue) for obsessive–compulsive disorder, announcing “a crisis of access”. This report, published in Nature Medicine, concludes: “DBS should not be considered a therapy of last resort in the therapeutic armamentarium, but instead should be considered as part of a sequential synergistic approach to complement the effects of conventional treatments.” The report then goes on to complain that the insurance companies won’t pay for it. Here is the lead example cited for efficacy: “16 patients were randomized into a two-week crossover period…During the crossover phase, the mean difference in Y-BOCS score was 8.3 points (P = 0.004); that is, an improvement of 25%”. Yikes! 25% improvement for 2 weeks?? Nature Medicine??
In all, 72 patients across 6 studies using 5 different stimulation sites were studied for mere weeks. The report states: “Long-term follow-up studies have also documented the durability of response over several years”, but it does not elaborate. ‘Response’ was defined as a minimum reduction of 35% on the Y-BOCS” (a checklist of symptoms). Most of the reductions during the blinded phase were barely above that. The largest effects occurred during the open phase—that is, the uncontrolled phase subject to placebo effects (Table 1).
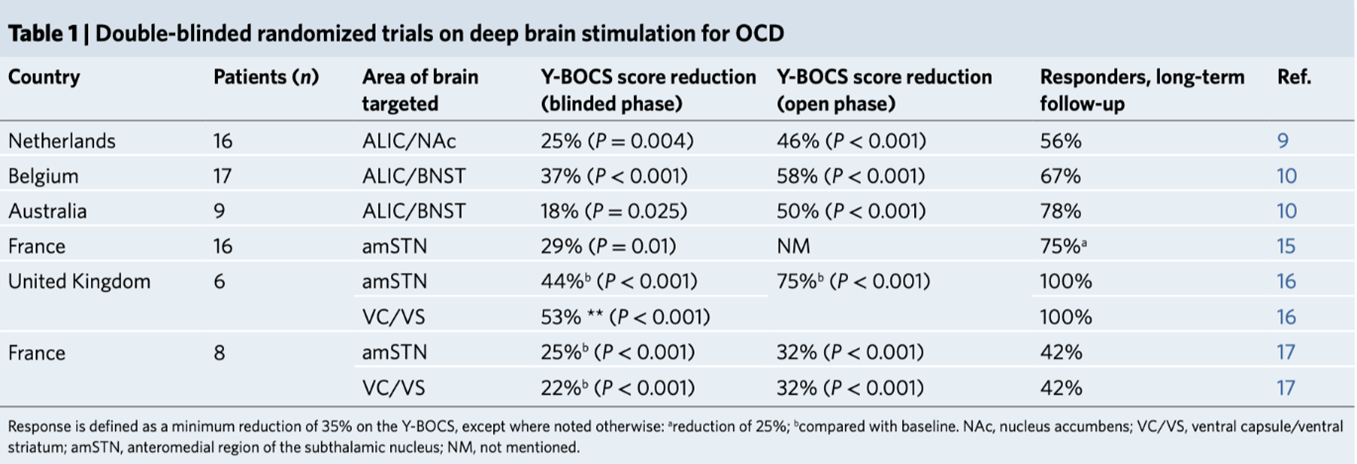
The lead author acknowledges membership on advisory boards and speaker’s honoraria from Medtronic, Boston Scientific, and Abbott, with a long list of similar “competing interests” for many of the other contributors. For Nature Medicine to publish such pitifully weak findings by authors with a financial stake in the industry strikes me as more evidence for a true “crisis”: rising collusion between Big Scientific Publishing and Big Pharma/Medical Electronics. This bouillabaisse of results, if written by neuroscientists studying rodents, would not be published.
Another single-case report used multiple implanted electrodes to treat depression and claims excellent results, apparently amounting to remission, for roughly a year (Sheth et al 2022). But consider these disclosures at the end of the article:
SAS is a consultant for Boston Scientific, Neuropace, Abbott, and Zimmer Biomet. NP is a consultant for Boston Scientific and Abbott. WG has received donated devices from Medtronic and is a consultant for Biohaven Pharmaceuticals. SJM has served as a consultant for Alkermes, Allergan, Axsome Therapeutics, Clexio Biosciences, Engrail Therapeutics, Intra- Cellular Therapies, Janssen, Neurocrine, Perception Neurosciences, Praxis Precision Medicines, and Sage Therapeutics. CCM is a consultant for Boston Scientific; receives royalties from Hologram Consultants, Neuros Medical, and Qr8 Health; and is a shareholder in the following companies: Hologram Consultants, Surgical Information Sciences, CereGate, Autonomic Technologies, Cardionomic, Enspire DBS. All other authors report no biomedical financial interests or potential conflicts of interest.
This report of a favorable short-term outcome for one individual was boosted by a Nature news article (Drew, 2022) that probably boosted stock prices for the authors even though history predicts relapse. Which brings us to Dr. Helen Mayberg.
Saga of Dr. Mayberg
Dr. Mayberg developed a conviction that severe depression is associated with pathophysiology in the frontal lobe. She performed some small, uncontrolled studies using DBS and persuaded a device manufacturer to sponsor a large randomized clinical trial (RCT). Based on poor early results, it was aborted. A long article defending Dr. Mayberg and claiming that she was badly treated by the RCT sponsors, was published by David Dobbs in The Atlantic (2018). Here, toward the end of the piece, is Dobbs’ admission:
Finally, Mayberg has long offered her patients comprehensive, personally tailored programs of psychiatric and social-service support to help them rebuild their lives. After years of deep depression, most peoples’ lives, relationships, and ways of thinking have been entrenched in illness and disability long and deeply enough that surgery alone was unlikely to address their whole experience. Like a patient with a knee repair, Mayberg said, “they need rehab to get well again.” So her team helps them get psychotherapy, occupational or physical therapy to rebuild skills or physical health, and other assistance to connect them to needed social services.
The broaden trial offered only postsurgical support, and to prevent statistical confounds it expressly ruled out the addition of any psycho- or drug therapy not underway before the trial. This successfully isolated the surgical intervention statistically—but likely reduced the chance of recovery.
In other words, Mayberg’s open trials seemed promising because of the support and charisma she provided, and when that was removed, the DBS itself didn’t work. Dobbs also notes that when the RCT was aborted and patients continued with DBS in the open condition, they gradually improved over months. But the claim for DBS is that the patients hop off the operating table good to go. So, the gradual improvement can be taken for a placebo effect—as in Mayberg’s own early work. This approach has now failed two randomized controlled trials (Drew, 2022; Dougherty et al, 2015)
The Review’s further proposals
The Review proposes new technology to better localize symptoms—such as those of depression, OCD, and mania—to specific brain regions, in order to visualize just the right spot to stimulate or coagulate. It even proposes that imaging can now help trace functional circuits for additional behaviors, such as “criminal behavior” and “free will”.
The Review misrepresents: (i) what current neuroscience actually knows about the functional architecture of primate prefrontal cortex and (ii) what current neuroimaging studies and genome-wide association studies (GWAS) reveal so far about “mental illness”. The Review ignores what recent longitudinal studies show about the prevalence and shapeshifting nature of mental disturbances. Each of these points is addressed below.
Functional architecture of prefrontal cortex [after Sterling & Laughlin, 2015, chapter 12]
All spikes carrying sensory information (except for olfaction), upon entering the brain, are routed to particular nuclei in the thalamus where they are processed and relayed forward to the cerebral cortex. There the signals are transformed into compact representations and sorted for further processing by specific areas for color, motion, objects and so on. These areas connect in distinct streams, for example, one visual stream identifies what and a different stream identifies where. All streams project forward—toward the front of the brain.
These processing streams gradually transform sensory patterns into abstractions. For example, early visual areas respond to oriented patches of bright and dark, later areas (in primates) respond selectively to faces and ultimately to particular faces. There are rich efforts to understand how this works; that is, to learn what these areas really compute, but most investigators would agree that their causal model of face recognition is provisional, a work in progress. This spectacular effort at tracing information flow employs multiple methods, including recordings from individual neurons within each identified area, recording signals of blood oxygen at millimeter spatial resolution and sub-second temporal resolution with supersensitive CCD cameras. This is the level of detail required to trace causal relationships relevant to human behavior. By contrast, the Review relies on methods whose resolution and sensitivity are less by orders of magnitude.
At the next deeper level of face analysis, there must be synaptic circuits to execute the hypothesized computations. But circuits for face recognition are not yet studied at this level of detail. This is where neuroscience is headed and needs to head—opposite to the direction proposed by the Review. The Review suggests that new imaging of neurological damage correlated with changes in behavior and mental state can potentially add something. Maybe so, but the greatest insights from that approach were gained during the late 19th and 20th centuries, and the bits proposed by the Review are unlikely to add a whole lot to what was discovered by Broca, Wernicke, and subsequent neurological investigations summarized by Norman Geschwind around 1965. Many subsequent investigators contributed modestly with this approach—for example, by analyzing individuals with callosal section (“split-brain”). It’s fine to continue this tradition of comparing lesions to losses, especially now that imaging has greatly improved. But it’s unlikely to yield much fresh insight. The Review’s assertion to the contrary is without foundation.
The sensory and perceptual mechanisms represented in the two visual streams were optimized early in brain evolution and conserved. For example, human retina and primary visual cortex are nearly indistinguishable from those of macaque monkey and chimpanzee (Sterling 2020, Figs 4.7, 4.8,). But as certain computations grew more important to humans, the essential cortical areas expanded greatly, especially in the frontal lobe. For example, the “lip smack” area in monkeys morphed into 15 distinct areas for human language (summarized in Sterling, 2020). The human prefrontal cortex expanded absolutely over that of the monkey and chimpanzee, but also expanded in proportion to brain size.
Since cortical processing proceeds from back to front, the prefrontal cortex accumulates all the computed wisdom: past and present; internal and external; urgent and less urgent. The prefrontal cortex integrates all the streams to predict what we most need, what will likely be most rewarding, and the optimal plan. The prefrontal cortex arouses the determination and persistence required to execute. The prefrontal areas couple subcortically to the striatum, which harbors “teaching circuits” that reward success and punish failure. Between the prefrontal areas and the striatum run circuits for checking our work; circuits for self-critique; circuits for our “self-chatter“—the voice in our head (Kross, 2021). All our capacities to produce art and music, to imagine and build the Webb telescope, to plan a mass-shooting, or self-annihilate are computed/concocted in prefrontal cortex. So naturally, when we suffer behavioral disturbances, that is bound to be the broad neuroanatomical locus.
However, it does not follow that each set of symptoms has a discrete locus that can be treated independently. The 15 areas are arranged according to the design principle minimize wire (Sterling and Laughlin 2015, chapter 13). This places the most strongly interconnected areas closest together, thus saving both space and energy for computation (Figure 1). Ablation or stimulation of any one of these areas must strongly affect all the others, plus the critical subcortical areas to which they connect.
The Columbia-Greystone Project
Efforts were made during the late lobotomy era to localize the supposedly positive effects of lobotomy by removing bits of selected Brodmann areas—the Columbia-Greystone Project (1947). A summary written 70 years later by group of neurosurgeons recounts:
Twenty-four patients were operated: …the cortical tissue of the rostromedial frontal lobe was excised until the white matter was reached. In this particular study, the mass of gray matter removed depended on the indication for surgery: 30–35 g were removed from each side for patients with chronic schizophrenia, 20–25 g for patients with obsessive-compulsive disorder or depression, and 15–20 g for patients with a primary complaint of pain. After a few preliminary surgeries, the investigators believed that the greatest effect could be achieved by focusing on the removal of Brodmann areas 9 and 10….The surgeons often noted that the relevant cortical matter appeared grossly abnormal (firm, atrophic, yellow, and/ or containing abnormal-appearing vasculature); however, microscopic tissue analysis did not show any consistent abnormality in the excised tissue. (Holland et al, 2017; my bolding)
The supposed abnormalities of the excised brain tissue reported by the original neurosurgeons were not confirmed by subsequent histology. We may conclude that these were not bias-free observations, but something closer to hallucinations. In the end, only 11 patients were well enough to leave the hospital, but of these, nine were rejected by their families, suggesting that they remained disturbingly disturbed. The Columbia-Greystone evaluations were entirely unblinded, so their final conclusions regarding who was helped and how much are almost certainly overstated. The 2017 authors (neurosurgeons) betray their own impression that these results were good, writing: “Despite the findings of the Columbia-Greystone project, psychosurgery practices began to decline significantly in the 1950s”. These modern neurosurgeons apparently accept uncritically the nonblind trial on 24 hospitalized patients subdivided into diverse small groups for different surgeries to treat diverse symptoms. These modern surgeons apparently believe the results were good enough to warrant continuing!
But in fact, these surgeries did not “work”. How could they? There was no rationale based on theoretical and experimental understanding of how the prefrontal areas work, nor what to expect when a surgeon removes 30-35 grams of brain tissue. What was supposedly pathological and how would that be “fixed” by removing it? Which area—when we know that they are all richly interconnected—would be “best” to remove? This bunch of neurosurgeons even today remain unconcerned—and so they represent a menace.
So-called “biomarkers” for mental disturbance
Frank disturbances at the level of local circuits may be manifest in sensory areas as a “spreading depression” for example, the “picket fence” seen during a migraine attack in V1. And when disturbed firing spreads across the motor cortex, it is manifest as a grand mal seizure that causes violent muscular contractions. But there is no evidence that disturbances of mood or behavior are caused by cortical circuits that are depressed or seizing. Despite efforts to find “biomarkers” for mental disturbance, they remain elusive (Insel, 2022). Two recent large-scale neuroimaging studies find no evidence to distinguish depressed from normal individuals or even to distinguish the two populations (reviewed in Sterling, 2022). This suggests that the Review’s claims to identify regions of interest in depression, including subcategories, and “criminal behavior” and “free will” are artifactual. Such claims emerge inevitably in small studies: the message from neuroscience needs to go out: distrust small studies.
The Review frequently returns to a particular goal: “modulate the neuroanatomy”. We can certainly conveniently “modulate” it now with TMS and such, but in what direction? What neuroscience is there to suggest that zapping the prefrontal cortex repeatedly would relieve depression? None. But when clinicians protest that “it works”, there’s the same old belief in snake oil. Most likely the benefits reported in small, short-term, poorly controlled studies will be positive. But over the longer run and larger studies, TMS won’t help because there’s not the slightest rationale. To mobilize efficaciously the frontal lobe circuits for mood, try a handshake, a touch, a smile, a hug, some praise, a walk. Not just once but daily—in place of the daily pills. Of course, this may not do for all, but it’s a start, and at least there is a strong rationale.
At a certain point, the Review pulls another fast one: it analogizes its therapeutic program to that of Wilder Penfield, who used electrical stimulation to localize and map various functional areas. The Review claims this approach can identify areas that cause mental symptoms for treatment by “localized” stimulation. But Penfield localized areas of normal function in order to avoid them while excising demonstrably damaged cortical tissue. Thus, when Penfield’s electrode encountered a “language” area in the left hemisphere, he recognized it because stimulation, far from improving the language function, interrupted the speaker in mid-sentence. This makes perfect sense because the neural computations required to comprehend and express language depend on cortical microcircuits that are far too complex to be “improved” by blind application of electrical stimuli.
The proper use of “model” organisms
Much great science has been accomplished by studying “model” organisms, including bacteriophage (virus that preys on bacteria), E. coli, the roundworm, C. elegans, zebrafish, mouse and nonhuman primates. A model is most useful when the level at which it is studied was evolutionarily conserved due to its success. Thus, our DNA replication is phage-like; our carbohydrate metabolism, protein synthesis and circadian clocks are bacteria-like; our G proteins, metabolic and developmental signaling are worm-like. Our nerve impulses are squid-like; our synaptic mechanisms are squid-, fly- and frog-like. Importantly for psychiatry, our neural circuit for reward learning is worm-like. That makes it half a billion years old.
On the other hand, when we employ a model organism to study phenomena that evolved tens of millions of years after our last shared ancestor, the comparisons can be misleading, even ludicrous. Thus, we have much to learn by studying the “connectome” of the mouse cerebral cortex because our brain has conserved much of the structure at the local circuit level. But for the next step up, the grouping of circuits into computational units, such as “columns” and “stripes”, and “blobs”, and their groupings into streams of specialized areas and “patches” to recognize objects, faces and linguistic symbols, the mouse cortex would have been not much help. The rodent’s “barrel fields” have been useful as a model of low-level computation and synaptic plasticity, but knowledge of the barrel fields would not have led us to imagine the functional architecture of the human visual system—that required concerted studies on non-human primates, whose structure we conserved for ~30 million years beyond our last common ancestor (Figure 2).
Now the rodent-psychiatry-industrial-complex claims the mouse as a model to study and treat mental disturbance, associated with the prefrontal cortex. But during the nearly 100 million years since our last common ancestor, our brain ballooned to ~3800-fold larger than the mouse brain (Figure 3).
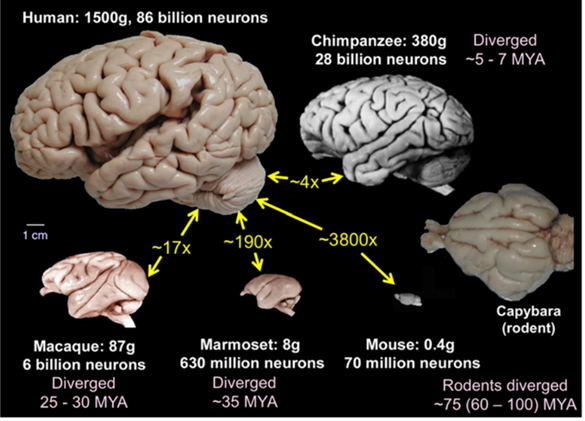
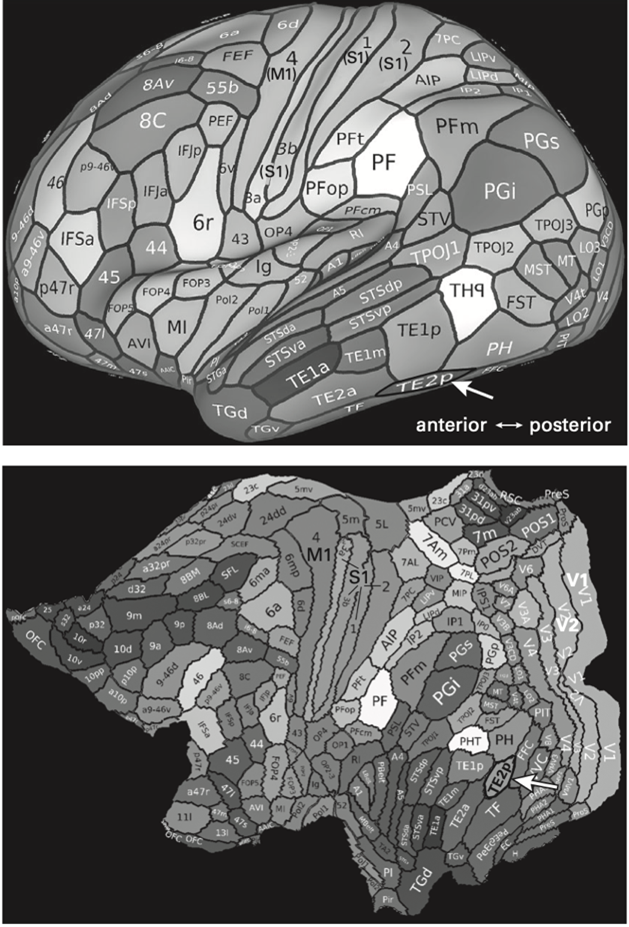
Moreover, whereas the mouse’s “prefrontal” cortex forms a narrow anterior crescent, like the pale margin on your thumbnail, the human prefrontal cortex, expanded disproportionately to occupy roughly one-quarter of the whole brain. The human prefrontal cortex includes about 20+ areas (Figure 4). None of these are present in the mouse brain, which therefore cannot serve as a model for their functional organization.
Regarding this grotesque comparison, one distinguished neuroscientist wrote to me: “I have a similar disdain for the rodent-psychiatry-industrial-complex. For example, it seems accepted that a mouse avoiding an open field is suffering from anxiety, when that’s the only intelligent choice if you happen to be a mouse. By analogy one might say that humans who refuse to swim across the English Channel are overly anxious. So let’s find a drug that makes them plunge in more readily…”
Cortical dendritic spines: a “biomarker” in schizophrenia?
This section grew rather long, but I am including it because the players are major leaders in our field, operating at the highest levels of the most esteemed institutions, and accessing the most prestigious journals.
Steven McCarroll of the Broad Institute gave an amazing keynote talk at a UMass Neuroscience colloquium in May 2019. He showed thrilling studies with new approaches to big genetic data. To introduce a slide about schizophrenia, he showed a spine-studded normal dendrite and a nearly naked ‘schizophrenic” dendrite (Figure 5) from the paper by Garey (1998) and reprinted by McCarroll’s colleagues in Nature News and Views (Johnson and Stevens 2018). I asked McCarroll about the image since it suggests very large differences—but he just cited that paper and and seemed unconcerned about its veracity. To me this suggested that McCarroll was invested in a story that whatever causes “schizophrenia” is associated with significant loss of dendritic spines. So, I investigated, and here is what I found.
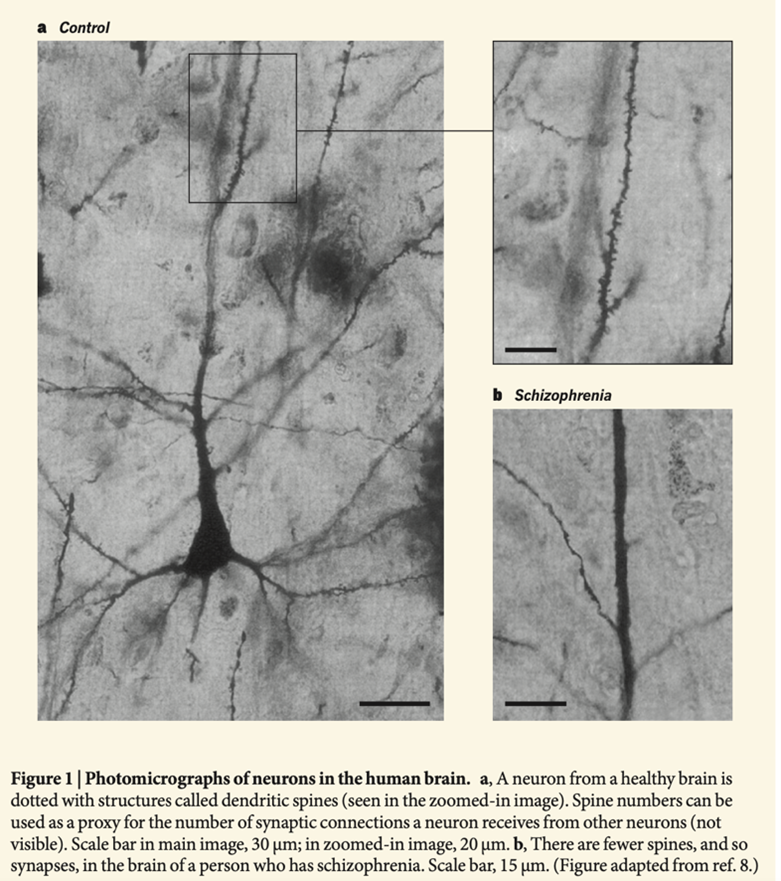
Prominent neuroscientists, including Eric Kandel (2018), assert that cortical spine density is reduced in schizophrenic brains in area 46—just where they’d like to locate the origin of the “disease”. These claims are based on Glantz and Lewis (2000) who used “Golgi rapid” impregnations of slices from this area on brains postmortem for more than 15 hours. The authors selected neurons to count spines on the basilar dendrites of pyramidal neurons in layer 3 (the apical dendrites were cut away). They reported statistical significance, with spine densities in schizophrenic brains lower by ~22%. A subsequent paper by Lewis and colleagues (Sweet et al 2009) reports similar spine reductions in auditory areas of postmortem brains whose owners had been diagnosed “schizophrenic” and suffered auditory hallucinations.
Glantz and Lewis speculate on how these differences might explain symptoms of schizophrenia and suggest spine molecular biology as a therapeutic target. Lawrence Garey (1998), using a similar method, reports a ten-fold larger difference. Lewis et al cite Garey, but not the order of magnitude discrepancy. The reprinting of Garey’s “schizophrenic dendrite” in the 2018 Nature News and Views paper also fails to mention the ten-fold discrepancy between the two studies. Thus, the hypothesis that someone’s disordered thoughts and sensory experiences during episodes of schizophrenia are caused by insufficient dendritic spines on these basilar dendrites of layer 3 pyramidal cells rests on three grossly discrepant studies of grossly deteriorated tissue.
Kandel’s book cites these papers as though they are true, illustrating the claim with an exaggerated drawing of the supposed difference between normal and schizophrenic dendrites. Moreover, Kandel locates the differences to apical, not basal dendrites as reported in the original papers. This is simply fake news—purveyed by a Nobel Prize winning psychiatrist with commercial interests.
Scientists who know lots about the genetic analysis, but who have never used the Golgi method, cite it as selecting neurons “randomly”—which is far from correct. The method is capricious and uncontrollable. Neuroanatomists know that certain neuron types appear frequently and other types rarely—far from their actual representation in the brain. When I consulted Javier DeFelipe (Director of the Cajal Institute) and an expert on cortical spines, regarding the reliability of the Garey and Lewis studies, given the postmortem delays, he replied: “In all sincerity, it is hard to believe those articles with the methods they used…”
It is disturbing to find that that this influential and widely-touted hypothesis (schizophrenia causally related to loss of pyramidal dendritic spines) rests on these three pitifully weak papers whose results differ by ten-fold. Why has this field—at the Broad Institute, for example—checked its critical faculties at the door? Later, I read David Linden’s technically excellent paper showing in mice that ovariectomy can reduce spines on cortical neurons by around 25% without obvious behavioral decompensations (Ye et al, 2019). And the report of DeFelipe et al (2008) with appropriate methods that human males show a 52% greater spine density in layer 5 than females and overall a 33% greater density. Apparently, spine density can be strongly modulated in various ways of which we currently have essentially zero knowledge.
Yet, Glausier and Lewis (2013) assert: “These [mental] impairments reflect alterations in neuronal circuitry within and across multiple brain regions that are due, at least in part, to deficits in dendritic spines…… Thus, while not conclusive, the existing evidence supports the idea that dendritic spine deficits are likely to represent the neural substrate for cognitive impairments in schizophrenia, and not to be a secondary consequence of the illness”.
Most likely, this spine-pruning hypothesis of schizophrenia will eventually dissolve because it arose from nothing. But what drove and sustained it in the first place? I suggest that it represents a determination to find a physical embodiment of mental disturbance and to grasp any straw that blows by. Maybe this straw is driven by the search for funds—it sounds good to have a specific hypothesis. But it maybe time for neuroscience to let this one go—along with the dopamine hypothesis, and the serotonin hypothesis…
Johnson and Steven Hyman (both at the Broad Institute where Hyman is Director of the Stanley Center for Psychiatric Research write in a 2022 review:
Pruning hypothesis comes of age.
The idea that disrupted pruning of neuronal connections in the brain during adolescence is a cause of schizophrenia was proposed in 1983. This proved prescient, as subsequent imaging, genetic and molecular research has shown.
But it did not prove prescient because toward the end of their editorial, they write:
Two foundations of Feinberg’s hypothesis appear unassailable: 1) that human neurodevelopment persists through the second and into the third decade of postnatal life and 2) that this same age range of late adolescence and early adulthood represents a period of remarkable vulnerability to schizophrenia spectrum disorders and other mental disorders (2). Successful investigation into the pruning hypothesis of schizophrenia is, in our view, dependent on better understanding the adolescent brain. Reframing the pruning hypothesis as a more general adolescent neurodevelopmental hypothesis would help resolve lingering debate regarding schizophrenia as a neurodevelopmental disorder: if brain development is not complete until after adolescence, then schizophrenia is neurodevelopmental, even if pathogenesis is largely explained by postnatal mechanisms (both genetic and environmental). More importantly, this framing emphasizes gaps in understandings of neurotypical adolescent brain development.
Notice that in all this verbiage, they do not summarize actual evidence that relates adolescent spine pruning to schizophrenia. This hypothesis is wafted forwards for reasons other than evidence. What restrained Johnson and Hyman from acknowledging the published 10-fold differences between different studies of human brains? It may be time to follow the money—by noticing in the Disclosures section: SEH [Hyman] has financial interests in Voyager Therapeutics, Q-State Bio-sciences, Emugen, Janssen, and F-Prime Capital.
Reprinted here is my 2019 correspondence with David Linden and McCarroll which (i) expands on Linden’s doubts regarding the hypothesis and the spine counts; (ii) shows McCarroll’s acknowledgement that he should not be touting the Lewis et al studies.
David Linden:
I share many of your concerns about links between pyramidal neuron spine density and schizophrenia. There is a well-established literature showing that, at least in rats and monkeys, spine density in hippocampal and some neocortical pyramidal neurons fluctuates by about 25-30% over the estrus cycle and yet the cognitive and affective changes that accompany these changes are subtle. Similarly, ovariectomy in female and castration in male rats, mice and monkeys all produce widespread pyramidal neuron spine loss of about 30% that is reversed by exogenous E2 or T respectively. Again, the cognitive and affective changes are subtle, arguing against a simple relationship with spine density. (my bolding)
And of course you’re right about Golgi reaction being non-random. Perhaps the best example of this is the unipolar brush cell of the cerebellum which has a crazy, unique shape, yet was completely missed by Cajal, Golgi, Lorente de No, etc. because it reacted poorly with the Golgi stain. It took Enrico Mugnaini using calretinin antibodies in the 1990s to find it. (11/04/19; edited for brevity)
Steve McCarroll:
Dear Peter and David,
I am enjoying this correspondence and must say that I agree with both of you. The field was too quick to make that image from the Lewis paper into an iconic image of schizophrenia pathology. (And I for one could stop contributing to that in my own seminars.)
Our data on C4 are consistent with many potential biological interpretations; the focus on the “adolescent pruning” interpretation is I think premature given evidence of kind David Linden has described and also the very real possibility that the classical complement pathway has additional neurobiological functions beyond pruning.
And I also have come to think that the focus on prefrontal cortex (e.g. BA46) has potentially been premature – yes its function is impaired in schizophrenia, but so are other brain functions, and we should be more thoughtfully considering the possibility that pathology starts in other brain areas (including thalamus, striatum and/or myelin) and percolates into other dysfunctions via alterations in circuits.
If there is one mistake that science can make over and over again, it is trying to connect the dots before we really know enough to see what the right connections are (11/04/19).
And yet, two years later McCarroll’s group (Yilmaz et al, 2021) report that overexpression of C4A “reduced spine density”. In their figure 5 (below), panel g indicates that the differences are rather small, especially considering their importance to the hypothesis, but the Discussion asserts a connection between behavioral changes that they compare to human schizophrenia and the supposed “pruning”.
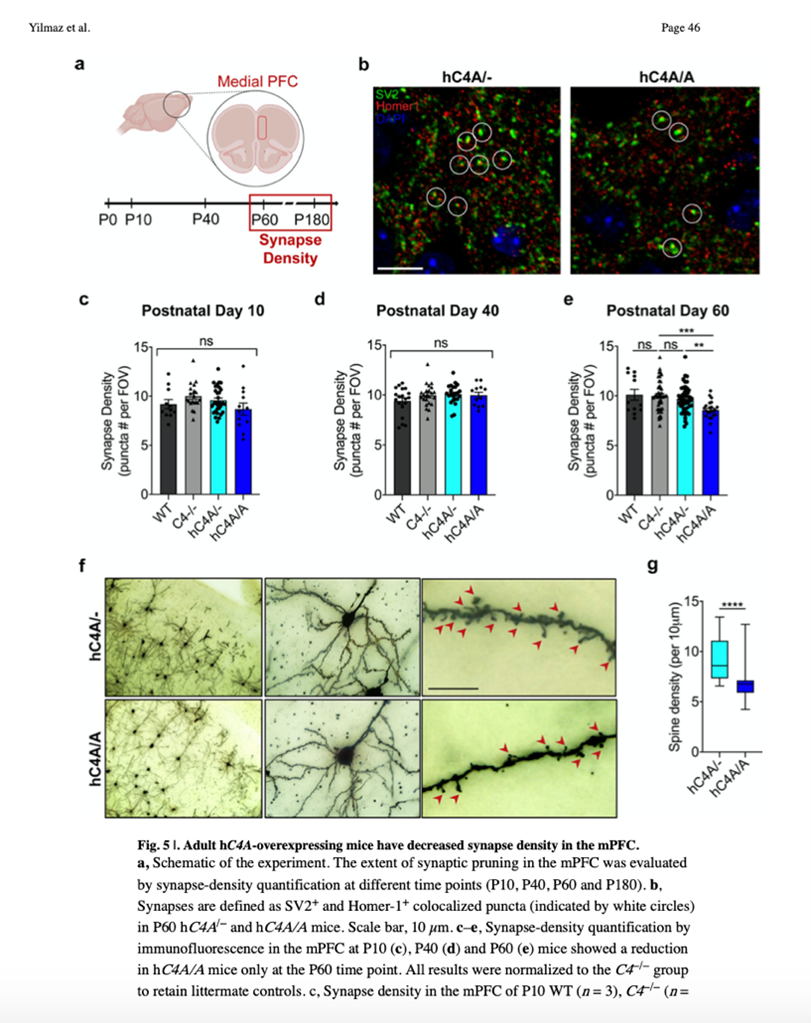
From the paper’s discussion:
Thus, our data suggest that the excessive elimination of synapses after C4A overexpression has pathological consequences on brain development, whereas C4 is not necessary, or has compensatory pathways, for normal brain development in the mPFC. Therefore, the use of therapeutic strategies targeting C4 downregulation can prevent and address the abnormal physiology resulting from C4A overexpression, while not distorting the endogenous synaptic refinement processes in the brain.
In short, despite McCarroll’s private admission of the weakness of the pruning hypothesis, in print he asserts it unreservedly and suggests therapeutic strategies. The paper continues to reference the bogus Lewis papers.
I understand that McCarroll and the Broad do amazing work. I do not oppose GWAS or molecular investigation of genetic contributions to mental disturbance. But I object strongly when prominent neuroscientists promote theories far beyond what is justified by their data and then start talking about therapeutic strategies. I have witnessed this across my whole professional life, so forgive me if I suspect, lurking in the background, a company with venture capital.
A large GWAS (Schizophrenia Working Group, 2014) identified a “risk locus”, the DRD2 dopamine receptor that binds anti-psychotics. But it also identified many more (totaling 108), including glutamatergic loci, v-gated Ca channels, and variants for synaptic plasticity. Each common variant associated with a mental disorder accounts for a very small part of the total variance. So, even if McCarroll et al are correct, that upregulation of C4A does modestly reduce spine density—well within known variations—do we expect it to “cause schizophrenia”—or to contribute a tiny bit to the probability? McCarroll and colleagues (Yilmaz et al, 2021) leave the impression that the role of C4A is causal and nothing minor about it.
A brief personal history of psychopharmacology and ECT
In the 1960s-70s, as “antipsychotic” drugs were introduced, one of the discovered pharmacological effects was antagonism of dopamine receptors (there was also histamine antagonism, but that was largely ignored). So, psychiatry invented the “dopamine hypothesis of schizophrenia”—too much dopamine—therefore “treat” by dopamine antagonists. But though the drugs suppressed certain aspects of mental disturbance and facilitated “management”, they worsened other aspects and caused permanent brain damage. By the late 1970s, 25-50% of patients in state hospitals were suffering from permanent tardive dyskinesia. Again, I testified in Federal District Court, and this time…we won! (Sterling, 1979; available at https://www.researchgate.net/profile/Peter-Sterling-3/research)
This victory was quickly overturned by the Court of Appeals which ruled simply, after just minutes of testimony, that to allow the verdict to stand would be to engage the Federal courts in every psychiatric drug decision. Gradually psychiatry crept away from the dopamine hypothesis (Insel, 2022), but it continues to prescribe “second-generation anti-psychotics” that suppress some mental disturbance but also derange metabolism causing huge weight gain, lethargy, type 2 diabetes and so on. To be clear, current prescribing of “anti-psychotics” is without any neuroscientific rationale. It is simply an empirical treatment that should be judged on its benefits vs. costs (long-term as well as short-term) without pretense of justifying these drugs based on neuroscience.
Starting in the 1970s, depression, anxiety and obsessive disturbances were treated with benzodiazepines, but they proved both addictive and lethal when mixed with other substances, such as alcohol. By the late 1980s, these dangers were obvious, and the patents were running out. Then it was that the so-called SSRIs (“selective serotonin reuptake inhibitors”) were introduced and promoted over benzodiazepines.
Here is Dr. Siddiqi tweeting to me (@whatishealth21) in 2022 to explain condescendingly that “benzos” are only prescribed by general practitioners, plus some nonsense about GABA resembling insulin.
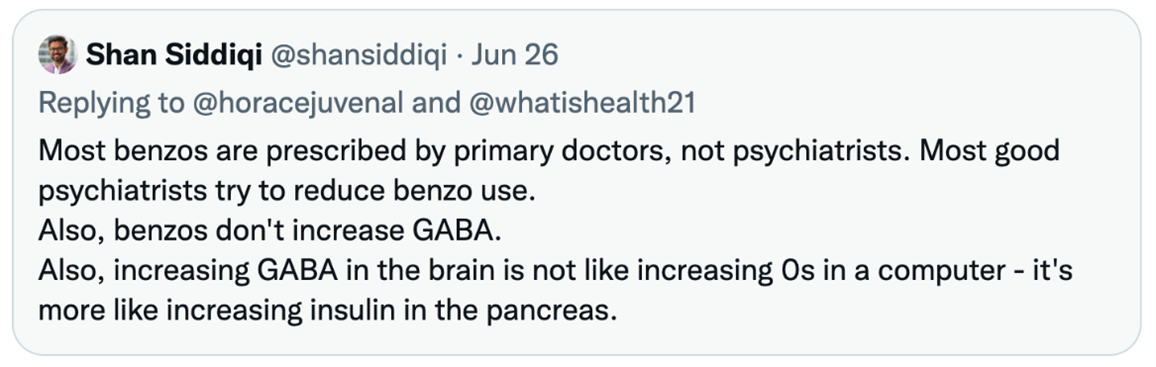
I confirmed his view of benzos with another senior psychiatrist, and it fits the pattern: promote a new “treatment” for the brain, and then sneak away as evidence surfaces of its inefficacy and iatrogenesis. The very psychiatrists who once touted ECT as utterly safe and effective now tout TMS in the same terms. Moreover, to promote TMS, they finally acknowledge that ECT really did quite a lot of damage. Damage was entirely predictable since ECT’s short-term effect on mood requires evoking grand mal seizures over multiple sessions, often followed by relapse and repeated sessions of ECT.
Here I would cite Suzanne Corkin’s study in the late 1970s of deficits that she correlated with numbers of ECT, and Irving Janis’ follow-up of ECT showing persistent memory losses (Sterling, 1978; Sterling, 2000). Reading Tom Insel’s Healing (2022), I was appalled that he fails to acknowledge that ECT, which he euphemistically terms a “reboot”, involves seizures that require a patient to be paralyzed by short-acting neuromuscular blocker, intubated and maintained on a respirator. Equally appalling is Karl Deisseroth’s Connections (2021), which uncritically claims that ECT is a therapeutic success. (For my summary of damage from ECT, see Sterling, 2000). Having testified at various state legislatures regarding the effects of ECT and publishing the above, I receive numerous inquiries from people who consider themselves “victims” of ECT. Naturally psychiatrists blame it on their depression.
Whether TMS will prove harmless over long run, we cannot know. For example, TMS is claimed to increase “synaptic plasticity”. Even if it were true, is that good over the long haul? Maybe there’s a reason why brain doles out its plastic moments carefully in neural time and space.
To sell the SSRIs, it was convenient to make up a story that depression, anxiety, and OCD are “caused” by low serotonin, because that is boosted by the SSRIs. Yet, the SSRI paroxetine (Paxil) affects not only serotonin but also dopamine, norepinephrine, and histamine. So, although paroxetine has been promoted as targeting serotonin, the brain doesn’t know this: it simply adjusts many transmitter systems (including 14 types of serotonin receptor) distributed all over the brain and in peripheral tissues and blood. To a drug salesperson, these are “side effects”, but to a brain already struggling with mental and behavioral disturbances, paroxetine must present quite a challenge.
After promoting SSRIs as preferable to addictive “benzos”, psychiatry and Big Pharma now acknowledge that they are addictive, though they term it “discontinuation syndrome”. Here is a communication to me from Prof G. Fava, retiring editor of Psychotherapy and Psychosomatics: “The switch from benzos to SSRI was purely commercial (no patent for benzos and patents for SSRI). It has been a spectacular achievement of propaganda, because there is no single study that indicates that SSRI are more effective.”
Then the story developed around MDD, so-called major depressive disorder. This disturbance is diagnosed with a checklist of symptoms with an arbitrary threshold—so many checks and you “have” MDD. This doesn’t rise to any sort of scientific reasoning that so many, since Francis Bacon, have worked hard to establish. MDD is just a bunch of shrinks making stuff up, as wonderfully clarified by Fried and colleagues (2022). Moreover, after some arbitrary number of attempts to help a sufferer, MDD is declared “intractable” or “treatment-resistant”, and that’s when the “treatments” get physical. The original descriptions by Freeman and Watts and all the old lobotomists tell the same story: Intractable. Off with the prefrontal cortex. I have friends who were depressed for 30 years and then recovered, so there’s no such thing as “intractable”. The ebb and flow of mental disturbance, including severe depression, is documented by Caspi et al (2020) in their large-scale, longitudinal study across four decades.
Elsewhere I review the failures of neuroimaging and genome-wide association studies to distinguish groups diagnosed as MDD from groups designated “healthy” (Sterling, 2022). Also cited is the recent review by Moncrieff and colleagues (2022) concluding that the transmitter depletion hypothesis of depression is unsupported and that antidepressant drugs offer most people little advantage over placebo.
Conclusions
(1) Current medicalization of all social and psychological suffering is unjustified by the currently known biology.
(2) Therefore, all the jiggering with human brains from childhood onward are wild empirical guesses—effects are claimed to “work” over a short time, and so are adopted and promoted for commercial reasons and for reasons of social control.
(3) Over my 50 years of following such treatments, I have observed that over the long term, none of them work, but meanwhile they cause considerable brain damage. Moreover, they deprive the sufferers of a sense of their own agency.
(4) By far the most potent measures to prevent and ameliorate mental “illness” and deaths of despair are social: stable housing for those who cannot manage without help; support for prenatal care and childcare; support for families, reduction of rampant childhood sexual abuse; education for young adults leading to decent work with decent pay; paid vacations; access to nature, such as neighborhood parks (South et al, 2021).
(5) The neuroscience community has been powerfully corrupted by dreams of glory and gelt. Notice that the guy we can’t trust with Twitter is the same guy who is preparing to hook our brains to his implantable device, Neuralink.
References
Alonso-Nanclares L, Gonzalez-Soriano L, Rodriguez JR, and DeFelipe J (2008) Gender differences in human cortical synaptic density. Proc Nat. Acad. Sci.105:14615-14619. Biological Psychiatry 92:246–251
Darby RR, Horn A, Cushman F, and Fox MD. (2018). Lesion network localization of criminal behavior. Proc. Natl Acad. Sci. USA 115, 601–606.
Darby RR, Joutsa J, Burke MJ, and Fox MD. (2018). Lesion network localization of free will. Proc. Natl Acad. Sci. USA 115, 10792–10797.
Deisseroth, K. (2021) Projections: The New Science of Human Emotion. Random House. New York
Dobbs D (2018) Why a ‘Lifesaving’ Depression Treatment Didn’t Pass Clinical Trials. April 2018. https://www.theatlantic.com/science/archive/2018/04/zapping-peoples-brains-didnt-cure-their-depression-until-it-did/558032/.
Dougherty et al. (2015) A Randomized Sham-Controlled Trial of Deep Brain Stimulation of the Ventral Capsule/Ventral Striatum for Chronic Treatment-Resistant Depression. Biological Psychiatry 78:240–248
Drew L (2022) Wiring up the brain to beat depression. Nature. 608: S47-48.
Fried EI, Flake JK, and Robinaugh DJ (2022) Revisiting the theoretical and methodological foundations of depression measurement. Nature Reviews Psychology 1:358–368.
Garey LJ, Ong WY, Patel TS, Kanani M, Davis A, Mortimer AM, Barnes TRE, Hirsch SR. (1998) Reduced dendritic spine density on cerebral cortical pyramidal neu- rons in schizophrenia. J Neurol Neurosurg Psychiatry. 65:446-453.
Glantz LA and Lewis DA (2000) Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch Gen Psychiatry. 57:65-73
Glausier JR and Lewis DA (2013) Dendritic Spine Pathology in Schizophrenia Neuroscience 251: 90–107. https://doi.org/10.1038/s41591-022-01879-z.
He W, Shao L, Wang H, Huang H, Zhang S, Li C, Zhang C and Yi W (2022) Bilateral Anterior Capsulotomy for the Treatment of Refractory Somatic Symptom Disorder: A Case Report. Front. Integr. Neurosci. 15:721833. doi: 10.3389/fnint.2021.721833
Holland R, Kopel D, Carmel PW, and Prestigiacomo CJ (2017) Topectomy versus leukotomy: J. Lawrence Pool’s contribution to psychosurgery. Neurosurg Focus 43 (3):E7
Insel T (2022) Healing. Our path from mental illness to mental health. Penguin Press New York
Johnson MB and Hyman SE. (2022). A Critical Perspective on the Synaptic Pruning Hypothesis of Schizophrenia Pathogenesis. Biological Psychiatry 92:440–442.
Johnson MB and Stevens B (2018) Pruning hypothesis comes of age. Nature554:439
Kandel E (2018) The Disordered Mind. Farrar, Straus and Giroux New York
Klyachko VA and Stevens CF (2003) Connectivity optimization and the positioning of cortical areas. PNAS. 100: 7937–7941
Kross E (2021) Chatter: The Voice in Our Head, Why It Matters, and How to Harness It. Penguin Random House New York
Kolluri N, Sun Z, Sampson AR, Lewis DA. Lamina-specific reductions in dendritic spine density in the prefrontal cortex of subjects with schizophrenia. Am J Psychiatry. 2005; 162:1200–1202.
Moncrieff, J, Cooper RE, Stockman T, Amendola, S, Hengartner, MP, and Horowitz, MA (2022) The serotonin theory of depression: a systematic umbrella review of the evidence. Mol. Psychiatry (2022). https://doi.org/10.1038/s41380-022-01661-0
Nils R. Winter, et al. (2022) Quantifying Deviations of Brain Structure and Function in Major Depressive Disorder Across Neuroimaging Modalities. JAMA Psychiatry. doi:10.1001/jamapsychiatry.2022.1780
Ofer N, Benavides-Piccione R, DeFelipe J and Yuste R (2022) Structural Analysis of Human and Mouse Dendritic Spines Reveals a Morphological Continuum and Differences across Ages and Species. ENEURO.0039-22.2022 1–14.
Schizophrenia Working Group of the Psychiatric Genomics Consortium (2014) Biological insights from 108 schizophrenia-associated genetic loci. Nature. 511: 421-438.
Sheth, et al (2022). Deep Brain Stimulation for Depression Informed by Intracranial Recordings. Biological Psychiatry 92: 246-251
Siddiqi SH, Kording KP, Parvizi J and Fox MD (2022) Causal mapping of human brain function. Nature Reviews|Neuroscience|23: 361-375.
South EC, MacDonald J and Reina V. (2021) https://communityimpact.pennmedicine.org/greening-for-health-equity-in-black-and-brown-communities/ JAMA Network Open. 4:e2117067
Sterling P (1978) Ethics and effectiveness of psychosurgery. in: Controversy in Psychiatry. eds Brodie and Brady. WB Saunders Philadelphia.
Sterling P (1979) Psychiatry’s Drug Addiction. The New Republic. December 8, 1979
Sterling P (2000) ECT damage is easy to find if you look for it. NATURE | VOL 403 | 20
Sterling P (2002) Comments on Brain Damage and Memory Loss from Electroconvulsive Shock. Testimony to the New York State Legislature. https://www.researchgate.net/profile/Peter-Sterling-3/research
Sterling P (2022) A neuroscientist evaluates the standard biological model of depression. https://www.madinamerica.com/2022/10/neuroscientist-evaluates-depression/#
Sterling P (2020) What is Health? Allostasis and the Evolution of Human Design. MIT Press Cambridge.
Sterling P and Laughlin S (2015) Principles of Neural Design. MIT Press Cambridge.
Sweet RA, Henteleff RA, Zhang W, Sampson AR, Lewis DA. (2009) Reduced dendritic spine density in auditory cortex of subjects with schizophrenia. Neuropsychopharm.; 34:374–389.
Valenstein ES (1986) Great and Desperate Cures Basic Books New York
Valenstein ES (1987) The history of lobotomy: a cautionary tale. Michigan Quarterly Review pp 417-437.
Veerle Visser-Vandewalle, et al (2022) Deep brain stimulation for obsessive–compulsive disorder: a crisis of access. Nature Medicine | www.nature.com/naturemedicine
Ye Z, Cudmore RH, Linden, DJ (2019) Estrogen-dependent functional spine dynamics in neocortical pyramidal neurons of the mouse. J. Nsci 39:4874-4888.
Yilmaz M, Yalcin E, Presumey J, Aw E, MaM, Whelan CW, Stevens B, McCarroll SA, and Carroll MC. (2021) Overexpression of schizophrenia susceptibility factor human complement C4A promotes excessive synaptic loss and behavioral changes in mice. Nat Neurosci. 24: 214–224. doi:10.1038/s41593-020-00763-8.



Dr Sterling,
Have you considered the fact that conditions like Bipolar disorder, PTSD, Borderline Personality, schizophrenia, ADHD, autism, addiction and depression all have high rates of MTHFR mutations that can cause nutrient deficiencies and alter methylation and neurotransmitter production?
Report comment
Undermethylation is a risk factor only. It is neither necessary nor sufficient to cause these kinds of presentations. Nor is treating Undermethylation necessary or sufficient to remediate them—I can attest to this personally, as my liver is methylating fine now after a round of supplementation and I still have nearly all my ‘mental health’ symptoms. Some other environmental or developments stressors have to be involved. With respect to physiological risk factors, PANS/PANDAS will serve just as well with or without liver Undermethylation for someone to present with emotional distress. But even then you’d have to explain how the immune system breaks down, so there’s be multiple variables involved. The immediate and developmental environments and all their psychosocial stressors would have to be involved. Moreover, attachment trauma and its epigenetic and developmental effects plus later life stressors are fully capable of leading to high levels of emotional dysregulation without under or over methylation being involved at all. And for ‘borderline personality disorder,’ upwards of 90 percent of people so diagnosed have disorganized attachment and severe childhood trauma, which beats out the correlations with methylation issues. 2/3 of addictions, moreover, can be explained through childhood trauma according to the ACES studies. I’m assuming methylation issues plus childhood trauma is a really bad combo that puts you at higher risk, especially since you’re likely to be subjected to iatrogenic harms in this case, but really neither Undermethylation or childhood trauma explain addiction fully, since stressors need to be present in the current environment. In each case, a complex developmental outcome like the presentations you’re talking about can never be reduced to one physiological cause, one environmental cause, or one developmental cause. This is just me talking about my opinion though, it has nothing to do with the article.
Report comment
Ryan,
Great answer, thank you. Actually, the MTHFR mutations is the risk factor, not the hypomethylation.. As a matter of fact if you look at the NIH page on drugs that cause nutrient deficiencies, it lists SSRIs and MTHFR mutations as a risk factor.
Why? Because both SSRIs and MTHFR mutations cause low Folate, Vit B12 and Vit D and increase homocysteine and oxidative stress. MTHFR mutations also cause lipid and purine abnormalities and SSRIs affect lipid and purine metabolism. As a matter of fact, Dr Lisa Pan found low Cerebral Folate, chronic oxidative stress, lipid and purine abnormalities in patients with treatment resistant depression.
https://www.nature.com/articles/s41398-023-02696-9
But iet’s see whether MTHFR mutations fit as a risk factor in the conditions you mentioned.
Addiction…studies have shown that low Vitamin D and Vit B12 can increase addiction and relapse and MTHFR mutations can cause both.
PANS/PANDAS …
Psychosis can be triggered by infection in people with MTHFR deficiency
https://bmcneurol.biomedcentral.com/articles/10.1186/s12883-017-0827-0
Studies have found that “MTHFR may contribute to the common pathogenesis of psychiatric diseases and that its variants may be essential in controlling the expression of psychosis-related genes.”
BPD….”attachment trauma and its epigenetic and developmental effects plus later life stressors”
As a matter of fact, a recent BPD study found that..
“Intense stress (first hit) produces vulnerability to depression in some cases (40%). Those who were vulnerable developed a depression-like phenotype after a second hit of stress.”
The vulnerability to depression resulted from “a persistent state of oxidative stress due to low brain-derived neurotrophic factor (BDNF) which controls the nuclear translocation of the master redox-sensitive transcription factor Nrf2, which activates antioxidant defenses.”
In other words, chronic oxidative stress and no way to stop it.
Studies have shown that the MTHFR C677T variant and childhood trauma predict depression because the variant causes limited oxidative stress defense.
https://pubmed.ncbi.nlm.nih.gov/23900311/
So I rest my case
Report comment
I am grateful that Peter continues to write and inform the public. I read everything I can find from him and recommend his book What is Health.
Report comment
“As practitioners of ECT switch over to transcranial magnetic stimulation (TMS), they finally acknowledge that it is “much safer”. That is, they finally admit that ECT is not safe. But TMS is just now taking off, so whether it is also unsafe may not be apparent for a decade.”
This is literally insane. Electro-Magnetism is a singular force. A pulsing magnetic field does exactly the same thing as a voltage differential. Both induce an electric current.
Report comment
Yes and all the reports of adverse effects of TMS to the manufacturer and FDA are the same as the adverse effects of ECT. Big surprise. I just think of the Tom Riddle Voldemort scene in Harry Potter. I imagine a big reveal at the end of this TMS business where someone takes a magic wand a moves the letters ‘TMS’ around and it spells out ‘ECT,’ and everyone gasps!
Report comment
ECT = ELECTRO-CUTION TORTURE….
Report comment
Mental Disturbance linked with More general categories worldwide shuch as genitc and nutrition….
Report comment
Thanks for sharing your response to our work. Appreciate the discourse. We presented lots of data, but I understand there could be an alternative interpretation. I won’t go through all of the math about causality here, since that’s alert described in the paper and will let your audience read both perspectives and decide for themselves.
I also agree that the tweet you shared looks nonsensical out of context. Makes more sense when you look at it as a response to your post which had factual inaccuracies (e.g. benzos increase GABA, benzos are like adding more zeros into a computer). But I certainly didn’t mean to advocate for benzos – I hate benzos (ask any of my patients), and always try to stop them when I see a patient who got them from a previous doc.
Also in reference to that tweet, please note that “most” is very different from “all.” My statement was a fact, not an opinion. PCPs prescribe more benzos than all other doctors combined:
https://jamanetwork.com/journals/jamanetworkopen/fullarticle/2722576
Report comment
….when you “always try to stop them”, do you at least recognize that anything other than a long, slow taper off of “benzos” constitutes medical malpractice and medical neglect?
Cruelty to their victims is a key feature of current psychiatric “practice”….
Report comment
“The neuroscience community has been powerfully corrupted by dreams of glory and gelt.” Thank you for speaking the truth, Peter.
As to the “Causality in Mental Disturbance,” I’m pretty certain the answer is corrupted societal systems (banking, medical, legal, religious, never ending war, etc.) largely. But as to “mental disturbances” dealt with by psychiatry, I’m pretty certain those are primarily a result of iatrogenesis.
Given the fact that the ADHD drugs and antidepressants can create the “bipolar” symptoms.
https://www.amazon.com/Anatomy-Epidemic-Bullets-Psychiatric-Astonishing-ebook/dp/B0036S4EGE
And given the fact that the “bipolar treatments,” some of which are the same as the “schizophrenia treatments,” can create both the negative and positive symptoms of “schizophrenia.”
The medical industry all is taught in medical school that both the antidepressants and antipsychotics can create “psychosis” and “hallucinations,” via anticholinergic toxidrome. And the antipsychotics / neuroleptics can also create the negative symptoms of “schizophrenia,” via neuroleptic induced deficit syndrome.
https://en.wikipedia.org/wiki/Toxidrome
https://en.wikipedia.org/wiki/Neuroleptic-induced_deficit_syndrome
Psychiatry is really nothing more than an iatrogenic illnesses creating industry, that entered into faustian deals, to cover up the sins of other industries and governments.
Report comment
Excellent article, thanks.
I wrote about a “comeback” of psychiatric neurosurgeries in the mid-2000s: https://danielleegan.files.wordpress.com/2015/09/psychosurgeries_thismag.pdf
https://danielleegan.files.wordpress.com/2009/10/back-to-the-frontal2.pdf
https://danielleegan.files.wordpress.com/2009/10/vickys-brain.pdf
https://thetyee.ca/Life/2007/05/02/Psychosurgery/
And about DBS, articles from 2006-2017:
https://thetyee.ca/News/2006/10/26/DBS/
https://thetyee.ca/News/2006/10/26/DBS/
https://www.madinamerica.com/2015/09/adverse-effects-perils-deep-brain-stimulation-depression/
https://www.madinamerica.com/2018/01/brain-implants-spinning-trial-results-protect-product/
ECT, 2018:
https://thetyee.ca/News/2018/08/24/Shock-Treatment-Electroconvulsive-Therapy/
Report comment
Man oh man, the comments section under the ECT article scared the hell out of me. So many “loving” family members convinced that their son/sister/cousin is severely mentally ill, “sick in the head”, and that it is definitely “biochemical and not social or due to parenting”. These people demand sympathy for the fact that they can’t get the system to force treat their “loved one”.
Truly terrifying. Makes me think I lucked out being abandoned by my entire family many years ago.
Report comment
I don’t want to say you lucked out being abandoned by your family, that sounds horrible. And I can attest to the fact that having parents and family members who are absolutely unwilling to recognize their own abusive behavior and contributions to their ‘loved’ ones’ suffering, and who double down on the application of maximum duress and coercive control to ‘fix’ said loved one (likely the source of their loved ones suffering to begin with, like parents who beat their kids and then their kids act out more under duress and so they beat them more ad Infinitum) was the worlds worst mind fuck. Add in their ability to leverage a psychiatric system to legitimize their invasive manipulations, and the application by this system of measures that exacerbate suffering even further, legitimizing even more extreme and coercive measures, and you’ve got a horribly traumatic and constantly revictimizing situation indeed. Quite frankly I don’t know how I had any sense of self or self respect left by the time I extricated myself from the whole thing, I had just bought that this was all ‘necessary’ and everything was in my head. But then my grandma died and I got left a ton of money and all of a sudden my parents couldn’t apply duress and all of their threats and manipulations were hollow, and I started to wonder why subject myself to their constant abuse if I’m not being forced anymore? I cut them off entirely when it became clear that setting boundaries would eventually end up with my mother trying to find some way to get me declared incompetent and sue for conservatorship of my trust. Anyway I don’t know why I’m typing my life story here… suffice it to say your situation sounds horrible and you are right the involvement of toxic family members willing and able to leverage your freedom and autonomy at the drop of a hat in unprecedented ways for any way of living they dissapprove of is indeed also horrible.
Report comment
There is the statistic from a spanish speaking psychotherapists that says that 50% of minors in his office are there because of “parental issues” not minors’. He I think argues somehow that some therapists stopped seeing minors for that reason.
The parent needs psychotherapy not the child. And what they call narcicism is for the most part untreatable. Worse when it is not just narcicisism but what they call psychopathy.
And some psychotherapists mention that usually the more aware about the familial “dynamics” is actually the victim/patient. He or she “works” at a higher level than the rest of the family/victimizers.
Report comment
Hi Danielle
I am a recipient of a psychiatric neurosurgery – I had an bilateral anterior cingulotomy in 2001. I have just finished a memoir, which corrects my supportive views of what happened, when I originally wrote about it (‘Life After Darkness; a doctor’s journey through severe depression’ published 2006).
I was still in the spell of traditional and not so traditional bio-medical psychiatry then. I had a transient recovery before going in for round two of depression.
I wish I had known at the time, but then again, I was so drugged up, had had over 100 ECT and then this charismatic, caring psychiatrist convinced me that psychosurgery was the answer. I can’t undo what happened. I just don’t want it to see others harmed.
Report comment
One of my closest friends had several rounds of “ECT”, or “ELECTRO-CUTION TORTURE” as I call it. That’s only the smallest tip of that iceberg of a life story. She suffers daily BECAUSE OF the so-called “treatment” of the local “community mental health center”….
I’m writing because I wonder if you want to swap emails, & work on actually DOING SOMETHING CONSTRUCTIVE to change a truly genocidal “medical-industrial-financial-complex”?….
I’m going to see if I can find your book on Amazon, etc.,….
Maybe that “psychiatrist” was actually TOO “charismatic”, and nowhere near “caring” enough?….
Please consider sharing your story here at MiA….
Most of MY story is in my comment history here!….
Report comment
Have you looked at enactivism as a cognitive science guiding neuroscience instead of last century’s cognitive science which was based on the brain processing information?
Report comment
Neuro-psychiatry is just an elaborate way for psychiatrists to distract themselves from facing emotional reality.
Report comment
“Birdsong” is just a beautiful way for victims of the pseudoscience drug racket of psychiatry to reclaim their human dignity, and to express the ugly truths they have survived so well so far….
Report comment
Sometimes I wonder if the whole idea of hallucinations and similar symptomatic concepts misleads researchers studying the neuroscience of schizophrenia.
Medical science and neuropsychiatry describe everything with extremely complex vocabulary and complex research, but within that complexity, there are some simplistic metaethical assumptions that remain undocumented.
Let me pick a few words from your text as examples:
– Mental disturbances
– Symptom
– To treat
– Disorder
– Neuropsychiatric phenomena
– Therapeutic
– Healthy
– Unhealthy
– Sick
– Harm
– Pathological
These words indicate the reason for research and its goals, but unlike everything else that can be observed, these concepts cannot be directly seen or pointed to.
Instead, researchers point to natural objects like the brain and then assign classifications that cannot be proven true or false but are assumed to be something that must be changed or that changes something else in a desired direction.
Thus, these words inherently have a moral aspect. For instance, while pain can be observed, the question arises: Is pain good or bad? Is reducing pain harmful or beneficial?
While these may seem like minor details since everyone wants to eliminate painful experiences, the presence of moral vocabulary makes the science built around it subjective due to preexisting assumptions that are not scientific or objective in nature.
If one follows metaethics, there are several different standpoints:
1. Propositions involving these moral concepts do not have a truth value.
2. Propositions involving these moral concepts have a truth value, but it is subjective.
3. Propositions involving these moral concepts have a truth value, but they are always false.
Notably, metaethical emotivism considers these concepts as expressions of emotions and imperatives. Therefore, when speaking of symptoms of schizophrenia, one could replace words like “symptom” and “schizophrenia” with “Inner visions and possible deeds of those having them and behaving abnormally make me feel bad. I want you to prevent them!”
Consequently, while neuroscience research deals with observable phenomena, there is a subjective element of attempting to identify and understand something that may not actually exist, drawing conclusions that depend on the observer and are not inherently objective.
For instance, when looking at images of neurons with structures called dendritic spines, researchers not only seek to understand their behavior and quantity within the system but also frequently draw connections to moral classifications and viewpoints, treating those as existing things similar to dendritic spines.
Moreover, certain quantities of dendritic spines and their associated neurons are then classified as healthy or unhealthy based on those connections.
This situation bothers me deeply. While following philosophy and making notes, the entire medical science is based on the Naturalistic Fallacy (the idea of something being good is reduced to some natural property) and Hume’s guillotine (from claiming that something is bad comes the assertion that one should do something about that bad thing, but there is no “ought” from “is”).
Of course, medical language avoids explicit moral terms to seem rational, but it hides assumptions and comparisons about how things should be within medical terms.
The paragraph you provided from the discussion of McCarroll’s group (Yilmaz et al, 2021) paper includes terms that relate to the feelings of the perceiver rather than just the perceived object:
– Excessive
– Overexpression
– Pathological
– Therapeutic
– Abnormal (in this context, expressed as something that must be prevented)
– “Endogenous synaptic refinement processes” (in this context, expressed as something that must be preserved)
This kind of emotional logic might not have scientific truth value, or it could be false. However, because these words represent feelings, they have at least four other variable properties:
1. Does the feeling of goodness or badness differ depending on a person’s position?
2. Does the intensity of the feeling of goodness or badness differ among people? Furthermore, does the order of personal importance change for that?
3. Are the consequences of preventing or provoking a phenomenon that matters only as feeling as presumed?
4. Does the feeling of goodness or badness and the order of personal importance change after those consequences happen?
Only when all four variables hold constant values shared by everyone can that logic lead to an outcome that everyone would feel good about.
This may be the case with many applications of medical science, such as antibiotics and hemorrhoid treatments, but it is often not the case with psychiatric treatments due to an untold conflict of interest and conflicting emotions among all parties involved.
I agree that neuroscience is important and interesting, but to study it properly, one should not simply accept a diagnosis and presume that it can be researched as an existing object separated from everything else. Otherwise there is a high risk for those causal misconceptions.
For example, if one wants to research a single symptom like visual or auditory hallucinations, the first step should be making observations outside the field of neuroscience and outside field of medical science to identify general patterns of the phenomenon. Only after verifying the general pattern should one attempt to find the counterparts of that pattern within the brain.
Otherwise, one might not even realize that there are many different general phenomena that intervene and everyone of use can likely induce and test symptoms of schizophrenia, such as hallucinations.
For instance, after doing something requiring high concentration, like playing a fast-paced video game prolonged time, if one suddenly stops and lies down, closing their eyes, they might experience a brief real psychosis and see or hear things depending on the senses they used earlier. One could even compare this experience with the positive and negative symptoms of schizophrenia and confirm that it is likely the same phenomenon. Then, one could empirically test all the variables that affect that experience and begin to confirm those hypotheses using neuroscience.
However, could one find that connection if they are only seeking abnormalities and waiting for terms like “disorganized thinking and speech,” “hallucinations,” “reduced ability to function normally,” and “abnormal motor behavior” when normally no one uses those words in such cases? These terms are true, but they are also not true, as they are cherry-picked out of their causal context, and their word choice is not neutral but rather scary and negatively connotated.
Even if dendritic spines and dopamine did not have a direct causal connection with schizophrenia, there is possibly some connection that can be found by reframing the concepts and not focusing on abnormalities. The correct approach would be to forget how things ought to be and to attempt to describe a self-regulatory system of the nervous system and establish connections between changes in measurements, brain images, behavior and external stimuli.
I am extremely amazed if there isn’t increased dopamine activity when anyone is facing a severe threat and I am as amazed if that change is limited to the amount of dopamine and not all other neurotransmitters. Similarly, those dendritic spines should have some real reason for their existence unrelated to moral terms like sickness. That part is missing from every schizophrenia study. No system can be described without including all its components and only components existing in objective reality.
Report comment
Yours is a really great post.
In Re 🙂 of the first part of the last paragraph:
The brainstem, and particularly the reticular formation, has probably hundreds of small groupings of neurons that have a lot, maybe all of the neurotransmitters in the brain. The brainstem has a lot of connections within it and to other parts of the brain.
The functional connections are not well elucidated, the anatomical ones are, but without the functionality are difficult to use as explanation.
And if I am not misremembering, there is feedback, positive and negative between neurons with different neurotransmitters as message within at least the reticular formation. A lot of the symptoms and signs of neuroleptics actually to me point to the brainstem, even maybe the cognitive ones, but I would be drawing a line too far I think.
Even if the brainstem is or was thought to be involved in sleep, circadian rythm and attention. The old upward activating system. Even generalized epilepsy at some point was thought to be “centroencephalic” and thus related to the brainstem. It was not originated from the surface, that was the hypothesis, then they discovered tiny scars on the cortex on some patients, some malformations, etc., but.
And the brainstem is not only involved in sensation and motion of the head and its parts, it is involved in basic bodykeeping, and emotion… even perhaps perception since there is some processing within the brainstem of the signal, even if only is just splitting the signal and crossing it to the other side.
And going upwards maybe even intepretation 🙂
Report comment
Pushing my fantasy further:
Just by splitting the signal, what if I hear or see at different speeds my left and my right perception field and that is sensed by my reptilian brain but not by my higher brain, and that gives me fear from apparently nowhere?. Like I fell through the mythical time wormhole without being aware? Alice in rose glasses tumbling down the rabbit whole.
Those hypotheses could sound like the new research criteria inspirators.
Report comment
“Janne”, you really need to write a longer or shorter piece here at MiA….
The above is TLDR, for me right now, but I’m looking forward to returning to it ASAP….
Your writing here is like a map out of the psychiatric wilderness we are all lost in….
Yes, I do indeed intend that quite literally….
Report comment
Hi Peter
I found your article fascinating and disturbing. I am a recipient of psychosurgery. After seven years treated with combos of drugs, over 100 ECT and basically slowly dying under lock and key, I was persuaded that a bilateral anterior cingulotomy would help me. The proponent was a team (still in existence today) in Dundee, Scotland – the Advanced Interventions Service, which was then headed up by an extremely charismatic and kind character Professor Keith Matthews. He won me over, which in retrospect was as much to do with his compassion, as my desperation. I had the surgery, couldn’t remember what had been done but made a spectacular recovery on Day 8. However, Professor Matthews and his team could not take the credit. THey had already stated that the surgery was not curative and ‘responders’ did not respond before six-nine months. THey could give no explanation to my recovery. This is all documented in a memoir in which Professor Matthews wrote a foreword. However, within a year of publication, I was back in his hospital once again. Further ECT ensued and then I was told I would need high dose anti-depressants for life with the expectation of further serious relapses.
I have defied their prognoses. How? By taking myself off all medication and getting counselling /psychotherapy for the underlying childhood adversity (and re-traumatisation at the hands of psychiatry), which I had already identified as the cause of the very first ‘breakdown’. It’s a shame that it robbed me of so much life and hurt my family. Apparently it is too late for legal redress. So I have written another memoir, recently completed, but as yet unpublished. Perhaps you might find it interesting? I am well now, in fact better than I have been for decades, working full time as an Emergency Physician! My passion in life is to stop others from being caught up in the nonsense that arises out of fraudulent or non-existent so called research, peddled by members of my own profession. I fell for it and I consider myself extremely lucky to have some sort of life afterwards. Many others do not. It is a travesty.
Report comment
Cathy, when you wrote, “couldn’t remember what had been done,” do you mean that you suffered some form of post-surgical/anesthetic amnesia and, if so, could you please tell us more about that?
You see, I recall Prof. Howard Schubiner’s telling of cases where people’s chronic pain reportedly went away along with other memories in instances of anesthetic amnesia…
Many thanks for your fascinating comments above, and wishing you ever-improving health,
Tom.
Report comment
Congratulations on your second memoir, Cathy. I will be very interested to read it when it comes out.
I have many parallel experiences — ECT, TMS, decades of drugs and diagnoses that only compounded the original (unacknowledged) problems. I understand the feelings of loss. I think many people will be helped by hearing your story.
Report comment
Hey! I wanted to be an ER specialist!
But I thought I could not stand the long hours without sleep being the ER so “tense”.
Alas, I was a chronic, still, insomniac. And ended in a specialty training place where ER trainees slept better!, critically better. We couldn’t even eat, verbotten actually, because it caused us sleep.
Oh, the ironies. 🙂
Report comment
Neuro-psychiatry is the modern adaptation of Dr. Frankenstein’s lab.
“Frankenstein and the Dangers of Unrestrained Science”, by Seth Bontrager, @web.colby.edu
Report comment
Gracias por la información
Report comment
I find it more amazing every day away from being coercively controlled by my mother and psychiatrist that one can be diagnosed as being in denial if they resist the notion that they have a purely biological disease there is no evidence for the existence of. It is very odd indeed. And what of the known adverse effects on psychological well-being of coercive control? Nope — those are caused by the disease too, and the only solution is coercive control…by your abuser. How anybody improves in this system is beyond me.
Report comment
As a psychiatrist for 30 years I have to agree with Sterling’s views that generally psychiatry and psychotherapy are pretty ineffective. Mind you I only realised this after moving into ADHD – the stunning results and long term effectiveness of this treatment really unmasked how ineffective previous psychiatric treatments actually were. I have always thought the dopamine and serotonin hypotheses are unconvincing….and having realised that half or more of all side effects are actually the same as the physical symptoms of anxiety…..I have to ask cui bono, and it doesn’t appear to be the patients…..but really clearly psychiatrists, psychologists and anyone who works in fortress mental health.
Report comment
This comment does not appear to reflect the Dr. Helen Read:
“I’ve been reviewing my NHS activity over the past 2.5yrs, since I moved to a front line mental health Consultant Psychiatrist role. It seems that I have diagnosed 413 new cases of ADHD in clients who were referred to our service for other mental health conditions. The referrals queried depression, anxiety, possible bipolar, personality disorder – the usual range of mental health conditions. Almost all of these patients once diagnosed and treated with medication were able to be discharged from the service and to my knowledge are all doing well with no need to be referred back for anything.”
From
https://adhdconsultancy.co.uk/blog/dr-helen-read-talks-with-matthew-bellringer/
Report comment