

On December 23, the FDA approved a new drug for schizophrenia, lumateperone, which was said to be a “first-in-class” medication, suggesting that its mechanism of action differed from antipsychotic drugs in current use. This new drug, an article in JAMA Psychiatry concluded, “demonstrated efficacy for improving the symptoms of schizophrenia and a favorable safety profile.”
After the FDA gave its stamp of approval, the stock price of Intra-Cellular Therapies, the company bringing lumateperone to market, soared on December 23, rising from $12.44 to $36.51. The company’s valuation rose $1.3 billion that day, giving investors reason to hoist a glass of champagne.
In the company’s press release, Jeffrey Lieberman, the former president of the American Psychiatric Association, who was a lead investigator in the trials and also served as an advisor to Intracellular Therapies, told of how lumateperone was a welcome addition to the medicine chest for schizophrenia.
“Schizophrenia is a complex disease that severely impacts patients and their families. Effective treatment in a timely fashion can be game-changing for people with schizophrenia,” he said. “The efficacy and safety profile of Caplyta approved by the FDA offers healthcare providers an important new option for treating people living with schizophrenia.”
That is the general story being told to the public, with the JAMA Psychiatry article, which was published online on January 10, providing a scientific stamp of approval for the FDA’s decision. It was a novel drug that had proven to be a safe and effective treatment for schizophrenia. An editorial in JAMA Psychiatry summed up the possible therapeutic advance it represented.
“Although we do not know what the ultimate comparative advantages of lumateperone will be, it is encouraging and potentially exciting to see a new drug with novel pharmacologic properties progressing through the gauntlet of drug development and US Food and Drug Administration approval.”
A Scientific Charade
While that is the public story, an in-depth look at the “evidence” for lumateperone reveals that the clinical testing and approval of this drug is best described as a charade, a game of scientific pretense. The clinical trials, in their design, served as the study of a drug for use after chronic patients have abruptly quit taking their psychiatric medications, as opposed to a study of a “drug for schizophrenia.” And even in that context, the drug did not reliably best placebo in any of three studies.
Nor is the “novel” drug claim fully accurate.
There is nothing in the design of the lumateperone trials that is particularly new, or even surprising given the history of the testing of new drugs for schizophrenia and the commercial influences on that process. The clinical tests of lumateperone did not deviate, in any significant way, from the norm. However, the approval of this drug does provide an opportunity to see anew that the testing of antipsychotics for schizophrenia is, from a scientific perspective, a fatally flawed process, producing “findings” that tell little about a drug’s efficacy as a “treatment for schizophrenia.”
Indeed, the most interesting finding to emerge from the lumateperone trials provokes a surprising question: Why do the chronic patients abruptly withdrawn from all of their psychiatric medications and randomized to placebo improve, as a group, during the 28-day study period? That result runs contrary to all that is known about the emotional and physical difficulties that people experience following abrupt withdrawal of psychiatric drugs. Patients who abruptly stop their medications are expected to become worse, at least for the immediate period following withdrawal, and yet that was not true in this case.
A Drug Like Clozapine
Lumateperone is being promoted as a novel compound that will not cause the side effects that existing antipsychotics do. Lumateperone, wrote the authors of the JAMA Psychiatry article, “is a mechanistically novel investigational agent for schizophrenia. The mechanism of action is unique because it simultaneously modulates serotonin, dopamine and glutamate neurotransmission, the key neurotransmitters implicated in serious mental illness . . . In addition, lumateperone lacks interaction with off target receptors that may contribute to the adverse effects of other antipsychotic drugs.”
In other words, this new molecule is something of a magic bullet. It “modulates” the very transmitters said to be the cause of schizophrenia, but—if the JAMA Psychiatry report is to be believed—does not interact with other neurotransmitters that cause the adverse effects that other antipsychotics do.
This is reminiscent of the “breakthrough” claims made about Risperdal and Zyprexa when they were brought to market in the 1990s. However, as the protocol for Intra-Cellular’s phase III study that was used to gain FDA approval makes clear, lumateperone is quite similar to clozapine in its effects on dopamine and serotonin.
All antipsychotics block D2 receptors, which is understood to be the mechanism that reduces hallucinations and other psychotic symptoms. At a therapeutic dose, most antipsychotics block more than 60% of striatal D2 receptors. Yet, as the protocol notes, Intra-Cellular’s drug does so as well. The 120 mg dose tested in its phase II trial blocks 70% of striatal D2 receptors, while the 60 mg dose blocks 50%.
Although the blockade with the 60 mg dose is slightly lower than most antipsychotics, it is similar to the D2 blockade with clozapine. Clozapine and other “second generation” antipsychotics also block serotonergic (5HT2a) receptors, with clozapine—the most potent of the drugs in this regard—blocking 90% of these receptors.
At a 60 mg dose, this is precisely what lumateperone does. Lumateperone, the protocol states, is “similar to that of clozapine with full saturation of cortical 5HT2a receptors when striatal D2 receptor occupancy is relatively low.”
Researchers have identified the characteristic side effects that flow from a drug’s blockade of a particular neurotransmitter. Blocking dopamine and serotonin pathways, while credited as providing a therapeutic benefit, causes a host of side effects. A clozapine-like drug may not cause extrapyramidal symptoms as regularly as a drug that blocks a higher percentage of D2 receptors, but it is likely to be sedating and cause metabolic changes that lead to significant weight gain.
While the editorial in JAMA Psychiatry touted lumateperone as having “novel pharmacologic properties,” it also acknowledged that its primary mechanism of action, the blocking of dopamine and serotonin, was what existing antipsychotics do. “Whether its mechanism of actions is truly novel remains unanswered,” the editorial stated. “It remains possible that lumateperone is just another ‘me-too’ second-generation antipsychotic.”
The Elements of a Charade
The protocol
Lumateperone was tested in one phase II study and two phase III studies, each one lasting 28 days, with similar designs. The article in JAMA Psychiatry reported results from study 301, with a link to a pdf of the protocol.
In study 301, the participants were required to have been in a “stable living environment” for the previous three months, and while in that community environment, to have suffered an “exacerbation” of schizophrenia symptoms. They needed to have had a prior history of responding positively to an antipsychotic. And most notably, investigators needed to decide that they could be “safely discontinued from current antipsychotic therapy, mood stabilizers, lithium, anticholinergics, and antidepressant medication.”
There was a long list of exclusionary criteria. The study volunteers couldn’t be suicidal, couldn’t be substance abusers (of alcohol or other drugs), and couldn’t have “clinically abnormal laboratory values.” They also couldn’t be suffering from any significant “hematological, renal, hepatic, endocrinological, neurological, (or) cardiovascular disease,” and they couldn’t have had a “history of neuroleptic malignant syndrome.”
Those recruited for the study were admitted to an inpatient unit, and on the first day were abruptly withdrawn from all psychiatric medications. If the clinical investigators decided that it would be appropriate to titrate the withdrawal of medications, it could be done over a maximum of four days. Everyone had to be off all psychiatric drugs for at least three days before randomization.
At the end of the screening period, which could last up to seven days, the patients were randomized into the study if they scored 70 or higher on the Positive and Negative Symptom Scale (PANSS). This inclusion criteria would select for patients that were at least “mildly to moderately ill.”
In study 301, patients were randomized to a 60 mg or 40 mg dose of lumateperone, or to placebo. Investigators were allowed to prescribe lorazepam during the 28-day study for “agitation, anxiety or to aid sleep.”
The primary measure of efficacy was a change in the PANSS score from baseline to the end of study at day 28. The study was designed, the investigators reported in their JAMA article, to have the power to demonstrate an “effect size of 0.4,” which would correspond to a six-point difference between drug and placebo in the reduction of symptoms.
The study population
As can be seen, the inclusion-exclusion criteria were designed to enroll a select group of “schizophrenia” patients. Those enrolled came from stable environments in the community, had previously responded positively to an antipsychotic, didn’t abuse alcohol or other drugs, and were in relatively good health, even though they may have had years of exposure to antipsychotic medications. These criteria were being used to select patients who had a higher probability of responding to an antipsychotic than a random recruitment of “schizophrenia” patients, and, given their relatively healthy profiles, were at lower risk of suffering adverse effects.
Pharmaceutical companies regularly use inclusion-exclusion criteria to select a subset of patients from a diagnostic group that will be most likely to respond well to the drug. However, it is well known that because of this selection process, the results may not reflect how effective a drug will be in a “real-world” population of patients.
The most dramatic example of this “real-world” problem comes from antidepressant trials. Two decades ago, the NIMH funded two studies of antidepressants in “real world” patients, and in both studies, one of which was the large STAR*D study, response and remission rates were quite poor, and notably lower than response rates from commercially funded drug trials.
This is the first element of pretense in this trial of lumateperone, an element common to most industry-funded trials of psychiatric drugs. The trial used inclusion-exclusion criteria to enroll a subset of patients that could be expected to have higher response rates and fewer adverse events than a “real-world” set of patients. The study volunteers were not representative of a cross-section of “real-world patients,” yet the results are being promoted as proving efficacy and safety for “real-world” use.
Prior drug exposure
In study 301, the 449 patients randomized into the study were, on average, 42 years old, and they had been diagnosed 17 years earlier. At least 94% had been taking psychiatric medications prior to the study, with nearly two-thirds on lorazepam. The most common antipsychotics that patients were taking were quetiapine, risperidone, haloperidol, olanzapine, and aripiprazole. All patients had a history of exposure to antipsychotics.
As is well known, antipsychotics and other psychiatric drugs change the brain. They perturb neurotransmitter systems, and in response, the brain goes through a series of compensatory adaptations in an effort to maintain a “homeostatic equilibrium.” At the end of this process, former NIMH director Stephen Hyman wrote in a 1996 paper, the brain is operating in a manner that is “qualitatively as well as quantitatively different from the normal state.”
For instance, antipsychotics block D2 receptors. In response, the brain increases the density of its D2 receptors. Thus, anyone who is exposed to an antipsychotic for any length of time will have this unusually high density of D2 receptors, and this is just one of the many changes that will be induced by years of exposure to antipsychotics and other psychiatric drugs.
As a result of this drug exposure, lumateperone was not being tested in “people with schizophrenia,” as though their brains were now in their original “disease” state. The drug was being tested in people whose brains had been dramatically remodeled by their years of exposure to antipsychotics and other psychiatric drugs, and if the patients in this study shared any common physiology, it was the abnormalities—such as an increase in D2 receptors—induced by exposure to the drugs.
Everyone knows this. Yet this scientific elephant in the room is never discussed. Instead, the pretense is that a drug-washout design removes the effect of this prior drug use, and returns the study participants to a pristine “schizophrenia” physiology. This allows for claims that a drug has been tested for a “disease,” even though it is in fact being tested in a population of patients whose brains are “abnormal” because of their years of exposure to the drugs.
This pretense, it should be noted, permeates 65 years of clinical RCTs assessing the short-term efficacy of antipsychotics. As Lieberman and colleagues confessed in a 2017 paper, there has never been a “randomized, double-blind, placebo-controlled study” of an antipsychotic in medication-naïve patients. In other words, there is no good scientific record that these drugs are effective, even over the short term, in psychotic patients whose brains have not been changed by prior exposure to antipsychotics.
No placebo control
As is well known, there is the possibility that people diagnosed with a disease may get better without treatment, and that is particularly true for people given a psychiatric diagnosis. A placebo control is designed to provide a measurement of this natural capacity for recovery, with the understanding that in order for a drug treatment to be effective, the drug must provide better results than the “natural recovery” in the placebo group.
In this study, the abrupt withdrawal design produces a “placebo group” that is suffering from withdrawal symptoms, as opposed to “unmedicated” schizophrenia, and there is a lengthy record of research documenting the hazards associated with abrupt withdrawal of antipsychotics and other psychiatric drugs. These hazards include a worsening of psychotic symptoms, anxiety, fear, depression, inner agitation, mood swings, headaches, insomnia, vomiting, and a possible worsening of extrapyramidal symptoms.
Indeed, the common thought is that abruptly withdrawing chronic patients from all of their psychiatric medications would be clinical malpractice, given that it is known to put patients at great risk of deterioration.
The charade here is obvious. A drug-withdrawal group serves as a stand-in for a placebo control, which provides a “comparison” that could be expected to make the drug look good.
This telling fact is also regularly hidden from readers of the medical literature. In the JAMA Psychiatry article, there is no mention of the fact that the participants had been abruptly withdrawn from their psychiatric medications immediately prior to randomization; that telling detail is missing from both the “Methods” and “Discussion” sections of the paper.
A drug-restoration study
Given the abrupt withdrawal of all drugs during the screening period, the trials of lumateperone are properly described as “drug-restoration” studies. Those who were randomized to lumateperone were put back on the same type of drug—one that blocks dopamine receptors—that their brains had become accustomed to.
In clinical settings, this of course is the usual practice. If a schizophrenia patient has abruptly quit taking his or her medications and is now suffering from psychotic symptoms, the patient is quickly put back on an antipsychotic. One would expect that this drug restoration would help alleviate the symptoms that flared up following the abrupt withdrawal of the antipsychotic in the first place.
Regardless of expectations, the point is that the lumateperone trials, like most industry-funded trials that utilize a drug-withdrawal design, only provide an assessment of the possible merits of this drug within a specific clinical situation: If chronic schizophrenia patients abruptly stop all medications, what is the short term effect of putting them back on lumateperone? Over the next 28 days, do they fare better than those who are not put back on an antipsychotic?
The missing data
The clinical course studied in the lumateperone trials is this: Schizophrenia patients living in the community suffer some degree of exacerbation while taking their medications. They are then abruptly withdrawn from all their medications and randomized either to lumateperone or to placebo. They remain in this treatment groups for the next four weeks.
The study, in order to provide a clear assessment of that course, should present three PANSS assessments for the participants:
- Scores at initial screening
- Scores after abrupt drug withdrawal (immediately prior to randomization)
- Scores at the end of the 28-day study period
In the lumateperone trials, there was no use of PANSS at the initial screening. Instead, potential recruits were screened with the use of a scale (CGI-S) that quickly assesses “global” clinical status, and those who scored at least a four were seen as potentially eligible, in terms of their symptoms, to be in the trial. Yet, even these initial CGI-S scores, assessed before drug withdrawal, are never reported.
Thus, there is no information that details the symptoms of the patients before they had their drugs abruptly withdrawn. However, it is expected that the abrupt withdrawal will exacerbate the patients’ symptoms, at least to a small degree. This withdrawal-induced exacerbation then becomes the baseline measurement for assessing efficacy at the end of 28 days.
With this design, a study participant can end up being evaluated as having “responded” to the study drug even though he or she is worse off than when first screened for the trial.
For instance, in the lumateperone 301 study, the mean baseline PANSS score for the patients was 89. But what would have been their PANSS scores prior to the abrupt withdrawal? It is possible that they might have been only mildly ill, which would have produced PANSS scores in the high 60s or low 70s, with the abrupt withdrawal then pushing up their score to 89 at “baseline.” Twenty-eight days later, their PANSS scores might have dropped to the mid-70s, and they would now be categorized as having responded to the drug, even though they were now worse than at the initial screening.
If these studies were done to provide clinically relevant information, then the patients’ symptoms should be assessed using the PANSS scale at all three moments: initial screening, after abrupt drug withdrawal, and end of study. Then it would be possible to assess whether “improvement” on drug was an artifact of the drug-washout design. In addition, it would be possible to see how much harm had been done to the study volunteers by the abrupt drug-withdrawal design.
The concomitant use of a benzodiazepine
In the lumateperone trials, roughly two-thirds of the patients had been taking lorazepam before they were withdrawn from their psychotropic drugs and randomized into the study. However, following randomization, the protocol allowed for investigators to once again prescribe lorazepam to treat anxiety, agitation, and insomnia. More than 70% of the patients, in both the placebo and drug arms, were given this benzodiazepine.
The use of a second psychotropic obviously confounds the efficacy that can be attributed to the study drug. As an expert in clinical trial design testified in a civil case involving Prozac, the concomitant use of a benzodiazepine in the Prozac studies was “scientifically bad,” as it would “confound the results” and “interfere with the analysis of both safety and efficacy.”
Again, everybody knows this. The prescribing of a benzodiazepine in the lumateperone trials could help hide adverse effects and help diminish many of the 30 symptoms that are assessed with PANSS. And while the use of a benzodiazepine is certainly of clinical use when people have been abruptly withdrawn from all of the psychiatric drugs and are suffering from withdrawal symptoms, the pretense comes from ignoring that their use confounds the study results.
This pretense extends to the article published in JAMA Psychiatry. There is no mention of the concomitant use of lorazepam. One has to read the protocol and the online supplement to find that out.
The ethics of abrupt withdrawal
It is well understood, by doctors and patients alike, that abrupt withdrawal of antipsychotics and other psychiatric drugs can be perilous. It exposes the patient to any number of physical and emotional risks, and in regular clinical care, is never advised.
Yet, in the lumateperone study, patients could be enrolled if the investigator determined that they could be “safely” withdrawn from all of their psychotropic medications.
This “safe” phrasing is meant to provide an ethical cover for the study. But how was this supposed to be done? The protocol did not provide any method for the investigators to figure out who among the screened patients could be “safely” withdrawn from their drugs. Nor was any explanation given for why there might be a subset of chronic patients that can, in fact, “safely” withdrawal from all their psychiatric drugs at once.
Indeed, if there exists a subset of chronic patients who can safely be withdrawn from all of their medications, then why isn’t “abrupt drug withdrawal” seen as an option in regular clinical practice?
This is yet another element of the charade. The inclusion criteria provides ethical cover for an unethical act.
The bottom line
There was nothing particularly unusual about the design of study 301 or the other two clinical studies of lumateperone. But, from a scientific perspective, these studies cannot be said to provide a test of the safety and efficacy of this drug for “schizophrenia.” The best that could be said of the trials is this: In a population of chronic patients diagnosed with schizophrenia, with years of exposure to antipsychotics and other psychiatric drugs, did lumateperone provide a benefit following abrupt withdrawal of the drugs they had been on?
The charade is to pretend that the trials tested something other than that.
The Efficacy of Lumateperone
The remarkable aspect of the FDA’s approval of this drug, through a fast-track process, is that even in that scientific context, as a treatment for patients withdrawn from all of their psychotropic drugs, it didn’t reliably provide a benefit.
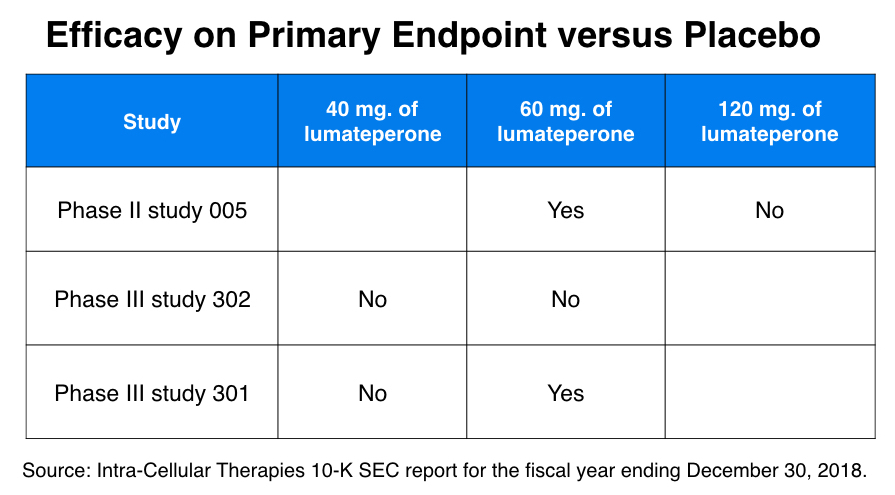
Intra-Cellular Therapies conducted one phase II study and two phase III studies as part of its application to the FDA. It provided summaries of these studies in its 2019 10-K filing to the Securities and Exchange Commission.
In the phase II trial (study 005), 335 patients were randomized to one of four treatments: 120 mg of lumateperone, 60 mg of lumateperone, 4 mg of risperidone, or placebo. The primary endpoint was reduction in symptoms on the PANSS scale from baseline to study end at day 28. While the 60 mg dose of lumaterone and the 4 mg dose of risperidone both provided a “statistically significant” benefit, the 120 mg dose of lumateperone did not.*
In one of the two phase III studies (study 302), patients were randomized to either a 60 mg dose of lumateperone, a 40 mg dose of lumateperone, risperidone or placebo, with reduction of symptoms on the PANSS scale the primary endpoint. Neither of the lumateperone doses “separated from placebo” on this endpoint, whereas risperidone did.
In the other phase III study, study 301, the 60 mg dose did show a “statistically significant” benefit over placebo on the PANSS scale, but the 40 mg dose did not.
Thus, in these three studies, there were six comparisons made between a lumateperone dose and placebo, and in four of the six, the drug did not provide a benefit over placebo. The 60 mg. dose was the dosage approved by the FDA, and in the two comparisons with risperidone, it provided similar results in the phase II study, but was inferior to risperidone in the phase III study.
Moreover, in study 301, the efficacy of the 60 mg dose over placebo was of a minimal sort. The baseline PANSS score for study participants was 89-90, and at the end of the 28 days, the average reduction in PANSS scores was 15.6 for the 60 mg group versus 12.4 for the placebo group.
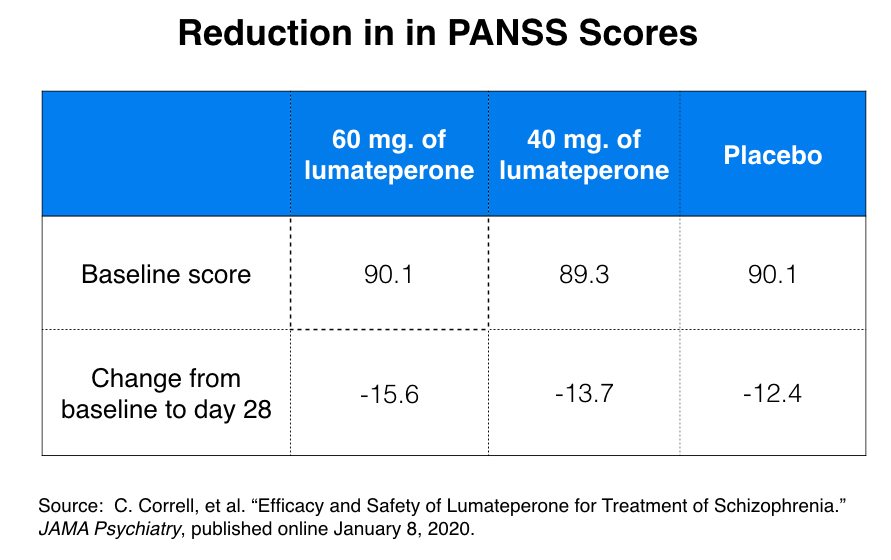 While this three-point difference was “statistically significant,” it is clinically meaningless. PANSS assesses thirty symptoms on a scale of 1 to 7, which means that scores can range from 30 to 210. In a 2012 paper, researchers determined that there needed to be a 15-point difference on the PANSS scale in order for that difference to be clinically meaningful. A three-point difference on a 210-point scale would not be clinically noticeable.
While this three-point difference was “statistically significant,” it is clinically meaningless. PANSS assesses thirty symptoms on a scale of 1 to 7, which means that scores can range from 30 to 210. In a 2012 paper, researchers determined that there needed to be a 15-point difference on the PANSS scale in order for that difference to be clinically meaningful. A three-point difference on a 210-point scale would not be clinically noticeable.
The study 301 investigators, in their JAMA Psychiatry article, also reported response rates, stating that a 20% reduction in PANSS scores would be evidence that a patient had “minimally improved” by the end of the study. Fifty percent of the patients in the 60 mg group responded by this measure compared to 38 percent in the placebo group.
This produces an NNT of 8, which means that eight people have to be treated with the drug to produce one additional “minimally improved” response. This also means that seven of the eight treated with the drug will suffer the adverse effects of the drug without receiving any benefit. These seven are composed of non-responders to the drug and those who would have responded without the treatment (the placebo responders).
Thus, in sum:
- There were only two instances—out of six tries—that lumateperone proved “statistically superior” to placebo on the primary endpoint of reduction in symptoms on the PANSS scale.
- In the positive 301 study, the superiority of the “effective” dose of 60 mg was of such a marginal sort that it was clinically meaningless.
- According to the “minimally improved” NNT of 8, seven of eight people treated with lumateperone will suffer the risks of adverse events without any benefit, as they will either be non-responders to the medication, or would have improved to the same extent without it.
The Safety of Lumateperone
Intra-Cellular Therapies, in its promotion of Caplyta, has touted it as safer and causing fewer side effects than antipsychotics in current use. The most common adverse reactions in the clinical trials, Medscape Psychiatry reported, “were somnolence/sedation” and “dry mouth.”
The JAMA Psychiatry article, in its conclusion, provided a similarly rosy picture of its side-effect profile. “The adverse events in the lumateperone groups that occurred were sedation, somnolence, fatigue, and constipation, which were all predominantly mild.”
This does seemingly tell of a great advance. No metabolic dysfunction, no dramatic weight gain, and apparently no extrapyramidal symptoms.
Here is how this safety profile was created.
List of common side effects
According to the study 301 protocol, the only “adverse events” that would be recorded would be those that newly emerged during the 28 days of treatment. The trial participants might have been suffering adverse effects from their prior years of psychiatric drug use, such as extrapyramidal symptoms, with such symptomatology remaining throughout the 28 days, but such symptoms would not be reported unless they newly emerged following randomization. In the JAMA Psychiatry report, these were described as “treatment-emergent adverse symptoms” (TEAS).
Second, the protocol was designed to minimize recognition of such adverse events. There was no use of an adverse-event checklist that would require the investigators to ask about potential negative effects. Instead, the protocol stated: “Adverse events may be volunteered spontaneously by the study subject, or discovered by the study staff during physical examinations or by asking an open, non-leading question such as ‘How have you been feeling since you were last asked?’”
This is a method that drug companies regularly use to minimize reports of adverse effects. If you don’t actively look for such negative effects, you won’t find as many.
This method produced the following list of common side effects, which seemed fairly benign.
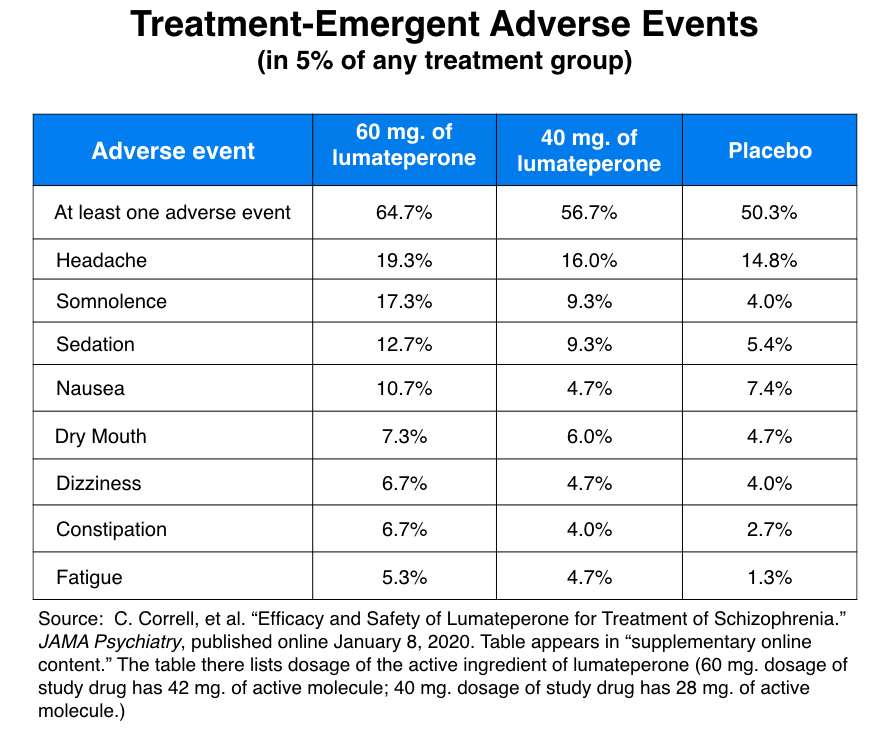
What is notably missing from this list is any evidence of EPS symptoms, and in their JAMA report, the study 301 investigators emphasized this point. “No EPS-related TEAS occurred in 5% or more of patients in any treatment arm,” they wrote. “EPS related TEAS were rare.”
Extrapyramidal symptoms
The first-generation antipsychotics—chlropromazine, haloperidol, and so forth—were notorious for causing extrapyramidal symptoms (EPS). Patients developed tics, dystonias, Parkinsonian symptoms, an inner agitation known as akathisia, and so forth. The second generation antipsychotics, such as olanzapine and quetiapine, were seen as improved drugs in large part because they didn’t block D2 receptors to the same degree as the first-generation drugs, and thus induced extrapyramidal symptoms to a lesser degree.
A drug that didn’t cause EPS symptoms at all, but was equally effective as existing drugs at improving PANSS scores, would be seen as a notable improvement. Although the adverse-event list seems to make the case that lumateperone doesn’t cause EPS, a company wishing to make this claim would need to show that it actively assessed EPS symptoms, rather than just waiting to see if such symptoms showed up as adverse events.
Intra-Cellular Therapies did this in its trials. The patients enrolled in study 301 were given baselines tests to assess the presence and severity of EPS symptoms and akathisia, and then these same assessments were made periodically during the trial and on the last study day. The investigators reported that based on these measurements, “treatment with (either dose) of lumateperone was not associated with increased EPS.”
The key word in that sentence, which can be easily missed, is increased.
In the JAMA Psychiatry article, the investigators reported on the mean changes in EPS symptomatology from baseline to day 28, as opposed to the percentage of patients who may have been suffering from such symptoms. Patients who were exhibiting extrapyramidal symptoms at baseline could still be exhibiting such symptoms at day 28, but as long as their symptoms hadn’t worsened, this wouldn’t produce any “mean change” in their EPS scores.
Furthermore, the investigators omitted the information that provided some insight into the percentage of patients who were, in fact, suffering from EPS during the study.
The protocol allowed for the prescribing of lorazepam during the trial to treat agitation, anxiety, and insomnia. All of these are EPS-related symptoms, with agitation and anxiety seen as cardinal signs of drug-induced akathisia (which often spikes following abrupt drug withdrawal). The investigators didn’t mention this fact in their article. But the online supplement does: more than 70% of the patients were prescribed lorazepam during the study for anxiety, agitation and insomnia.
Yet, these adverse events are missing from the list of general side effects.
Given this presentation of safety data, it’s impossible to know the percentage of patients in study 301 who were suffering from EPS. It appears that there were only a few instances when such symptoms newly emerged, and on the whole, there was no “mean change” of EPS symptoms in the patient groups over those 28 days. Yet, more than 70% of the patients were prescribed lorazepam to treat agitation, anxiety, and insomnia, symptoms that are a sign of akathisia. It would seem then that 70% or so were suffering from this “adverse event.”
The company, in its press release, stated that the “incidence of extrapyramidal symptoms was 6.7% for Caplyta and 6.3% for placebo,” which would indicate that its drug doesn’t cause such adverse effects. But information in the company’s press release and the JAMA Psychiatry article about the use of lorazepam in the studies is absent, and so the public is left in the dark about the potential for this drug to cause “agitation, anxiety and insomnia.”
Metabolic dysfunction
With the second-generation antipsychotics, metabolic dysfunction—weight gain, diabetes and so forth—moved to the forefront as the most problematic adverse effect. The study 301 investigators reported on this worry in the same way they reported on EPS symptoms.
The protocol required investigators to assess metabolic factors at baseline, and then again during the study and at day 28. But in the JAMA article, the investigators only reported “mean changes” from baseline to day 28, and thus if the patients had abnormal readings on metabolic factors at baseline, as long as metabolic-related measurements didn’t worsen during the four weeks, it would seem that lumateperone didn’t cause such problems.
As the investigators wrote in their JAMA article, “there were no significant mean changes in metabolic parameters from baseline to day 28 compared with placebo.”
Yet, just as the regular use of lorazepam in study 301 tells of patients who continued to suffer from symptoms that are linked to akathisia, weight-gain data published in the supplement suggests that lumateperone, like clozapine, may cause metabolic problems, at least for some patients. Twelve of the 143 patients in the 60 mg group had an increase of more than 7% of their baseline body weight in 28 days, which indicates that they gained 10 to 15 pounds (or more) in this short period.
The bottom line
It may be that lumateperone has a relatively benign safety profile compared to existing psychiatric drugs. However, what is clear is that the company and its investigators, in their collection and reporting of adverse event data, did not provide a picture of the presence of metabolic dysfunction and EPS symptoms in trial participants during the study. The fact that there were no “mean changes” in these domains indicates that at least those treated with lumateperone didn’t worsen in those two domains during their 28 days on the drug. But that doesn’t translate into evidence that lumateperone won’t cause such negative effects in some significant percentage of real-world patients.
The FDA’s Approval of Lumateperone
After the company completed its phase III trials, the FDA, in November of 2017, gave lumateperone its “fast-track” designation. This was granted, the company stated in a press release, because “lumateperone has the potential to address the unmet medical need for the treatment of schizophrenia with significant improvements on several clinically significant safety parameters, including with respect to metabolic, motor and cardiovascular issues associated with many currently available antipsychotic agents.”
Once the company filed its New Drug Application, the FDA scheduled an advisory committee meeting for July 31, 2019. But it cancelled this meeting on July 23, which spooked the investment community. Perhaps the FDA hadn’t found the lumateperone data very convincing.
Those fears were quickly quelled. On September 10, the FDA announced that it no longer planned to hold an advisory committee review, and three months later, on December 23, it approved the drug. It did so despite the lack of two positive phase III trials, which is the usual standard for getting a drug approved.
Earlier in 2019, the FDA approved eskatamine as a treatment for depression even though the efficacy evidence was of a similarly unconvincing sort, and with this approval of lumateperone, financial analyst Paul Matteis explained the pattern that was now evident. “The FDA is more flexible than average for neuroscience drugs” and is now taking “a glass half-full approach” to approving such drugs, he wrote.
The development of lumateperone did produce financial benefits for those involved. Investigators who conducted the trials were of course paid for this work, and as noted above, after the FDA announced its decision, Intra-Cellular’s valuation jumped $1.3 billion in a single day. Both Intera-Cellular’s CEO and CFO have sold shares of their stock since that day, each receiving more than $1 million from these trades.
Analysts said the company was expected to price Caplyta at $900 a month when the drug is launched in the spring. That is more than 200 times the monthly cost of generic risperidone at Walmarts or other pharmacies that sell generic antipsychotics at discounted rates. In this realm of commerce, the costs will be borne by those who purchase the pills.
The Unanswered Question
As mentioned earlier, there is one result from the lumateperone trials that warrants further investigation. Why did the PANSS scores of chronic patients who were abruptly withdrawn from their medications and then randomized to placebo improve over the next 28 days? Relapse studies regularly tell of withdrawn patients whose psychotic symptoms increase in the first month, and the general understanding is that abrupt withdrawal regularly leads to the eruption of many difficult symptoms. But not in this case, and such improvement in the placebo group is regularly seen in industry-funded trials, in spite of their drug-withdrawal design.
The improvement in the placebo group in this trial could be evidence that supports the regular use of tapering protocols. If chronic patients can improve following abrupt withdrawal of all of their psychiatric drugs, what improvement might they experience over longer periods of time if their psychiatric drugs were gradually withdrawn? However, another possible explanation is that during an industry-funded study the evaluators are expecting to see improvement in the participants, a bias that extends to both the drug-treated and placebo patients.
But that is not a question that interests commercial funders of drug trials or apparently the investigators in the lumateperone trials. In their JAMA Psychiatry report, they didn’t even mention the drug-withdrawal design. There is no discussion of this improvement in the placebo group, which represents yet another way that these trials do not help inform clinical care in any meaningful way.
*******
* The molecule that was tested in the trial was lumateperone tosylate. The 60 mg dose had 42 mg of lumateperone, while the 40 mg dose had 28 mg of lumateperone. In the JAMA Psychiatry article, some of the findings are presented using the 42 mg and 28 mg numbers.
MIA Reports are supported, in part, by a grant from the Open Society Foundations



“It may be that lumateperone has a relatively benign safety profile compared to existing psychiatric drugs”
Doubt it Robert, but very good work indeed!! It’s just more nasty crap, promoted by more bullcrap to make a mint. We need to find a way to inform these hedge funds that not only do they need to fund ethical sustainable energy, they need to stop funding/supporting products that only serve lies and terrible harms, violence and destruction.
They may have tested individuals CYP450 phenotypes to ascertain if they would get through the trial without becoming toxic. If you could find that out it would be very telling to their claim it doesn’t cause Akathisia.
The prescribing info suggests issues with CYP3A4 the sink enzyme which picks up everything the other enzymes couldn’t cope with:
https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/209500s000lbl.pdf
Report comment
PROTOCOLIZE RESEARCH REVIEW
Thank you for your review of this study.
You and other writers of Mad In America have gained crucial experience in examining research, consisting in tests and analysis criteria.
Isn’t it time to compile all these criteria in a protocol, allowing to carry out a systematic review of research according to predictable and rigorously defined criteria?
We always find the same criteria in your articles and those of other editors:
_ representative vs non-representative population
_ placebo study vs withdrawal study
_ naive population vs non-naive population
_ Confounding factors (including medication)
_ short term vs long term
_ protection of the safety (in particular due to withdrawal)
Etc.
There is not much missing to compile all these criteria in a systematic analysis protocol.
I would like different Mad In America editors to come together to establish such a protocol, which would guide the editors in their analysis of studies.
It makes it possible to assess the quality of studies in a stricter, more in-depth and better defined manner.
Such a protocol could eventually become a recognized standard for evaluating scientific research.
Report comment
? https://www.theguardian.com/world/2020/jan/19/isabel-dos-santos-revealed-africa-richest-woman-2bn-empire-luanda-leaks-angola
Report comment
Most scientific research today is not repeatable. Which means it’s trash. Asking MiA to make a difference (to the degree you suggest) is really not within the realm of possibility. Your best bet, if you have an ailment, is to do your own research. It’s essentially what Thomas Szasz was saying. “If you want a lobotomy, have a lobotomy”. Liberty …
Report comment
https://www.thejournal.ie/scientology-anti-psychotic-drugs-cchr-4959491-Jan2020/
“….THE HSE AND clinical psychiatrists have strongly criticised pamphlets being delivered across Dublin regarding antipsychotic medicines.
Leaflets warning people about the dangers of taking prescription antipsychotic medication were sent to residents in south Dublin by a group linked to the Church of Scientology in recent weeks.
The pamphlets are titled “Antipsychotics: The facts about the effects”, and are part of what is described as a psychotropic drug series published by the Citizens Commission on Human Rights (CCHR)….”
“…..The information contained within this leaflet is misleading, inaccurate and shows no evidence of any understanding of the complex biological, psychological and social contributors to mental illness. The HSE advises that if this leaflet causes concern to any person who is being treated with antipsychotic medication, that they contact their GP or a member of the mental health team that is looking after them. …”
Report comment
I read the quotes from the pamphlet in the article. It seems to be pretty straightforward, and the criticisms forwarded are generalizations and don’t appear to address any of the specific claims in the pamphlet at all. Saying something is “misleading and inaccurate” without saying what is inaccurate or misleading about it is a pretty lame criticism!
Report comment
I looked up the Pamphlet and I thought the descriptions were fairly honest.
Report comment
Hmm. Notice how astronomers don’t get their panties in a wad because Flat Earthers exist.
The Church of Psychiatry knows they’ve got the Good Stuff. They don’t take kindly to any one invading their turf.
I got suspicious of NAMI when I realized they were censoring what we could read or listen to. They canceled a speaker who got better off her drugs and wouldn’t tell us why. I found out from the grapevine. Then I realized the people running the show–the “normals”–were also listening in on our conversations.
They knew the truth would set us free. And it scared them.
The more hysterical rants I hear demonizing the anti-vax crowd the more distrustful of the flu vaccine I become too. 😛
Report comment
True enough. If the facts supported the pro-psychiatry viewpoint, why would they be so bent on suppressing any kind of dissent with these sleazy tactics? The only reason people viciously “attack the messenger” instead of addressing the concerns raised is because they know they have something to hide.
Report comment
Indeed they hide.
One thing they constantly try to stay on top of is their internal anger.
The most oppressed people I know. Before I knew something about myself, I had no idea how pervasive their sickness is.
I would like to be locked in a room with a shrink and wear him down. It should not take long.
Report comment
Thank you very much for this well done analysis. As a clinician this gives me the information I need when patients begin to ask about this drug. It is a shame that the drug companies get to do fraudulent research yet the clinician is obliged to follow along – unless articles such as this are published. Thanks again Robert, you and MIA are helping more people than you can ever know.
Report comment
Thank you for pointing out the scientific fraud rampant in the drug approval process, Robert. And I’m glad the clinicians who feel “obliged to follow along” are helped by your research, so they do not have to behave as lemmings.
But one does wonder how long it will take the psychiatrists to realize that all their antipsychotics/neuroleptics can create the negative symptoms of “schizophrenia,” via neuroleptic induced deficit syndrome. And the antipsychotics can also create the positive symptoms of “schizophrenia,” via anticholinergic toxidrome.
Report comment
Someone Else: This one is Quite Scary
“Schizophrenia: Early Warning Signs”
https://youtu.be/9JH7G1aekxc
Report comment
Does teeth extraction tests been performed for lumateperone?
Report comment
good question#, manyshev
Report comment
Robert Whitaker wrote: “As is well known, antipsychotics and other psychiatric drugs change the brain. They perturb neurotransmitter systems, and in response, the brain goes through a series of compensatory adaptations in an effort to maintain a “homeostatic equilibrium.” At the end of this process, former NIMH director Stephen Hyman wrote in a 1996 paper, the brain is operating in a manner that is “qualitatively as well as quantitatively different from the normal state.”
For instance, antipsychotics block D2 receptors. In response, the brain increases the density of its D2 receptors. Thus, anyone who is exposed to an antipsychotic for any length of time will have this unusually high density of D2 receptors, and this is just one of the many changes that will be induced by years of exposure to antipsychotics and other psychiatric drugs.
As a result of this drug exposure, lumateperone was not being tested in “people with schizophrenia,” as though their brains were now in their original “disease” state. The drug was being tested in people whose brains had been dramatically remodeled by their years of exposure to antipsychotics and other psychiatric drugs, and if the patients in this study shared any common physiology, it was the abnormalities—such as an increase in D2 receptors—induced by exposure to the drugs.
Everyone knows this. Yet this scientific elephant in the room is never discussed. Instead, the pretense is that a drug-washout design removes the effect of this prior drug use, and returns the study participants to a pristine “schizophrenia” physiology. This allows for claims that a drug has been tested for a “disease,” even though it is in fact being tested in a population of patients whose brains are “abnormal” because of their years of exposure to the drugs.”
That’s the bit when a lot of minds spontaneously shut-down. I struggled for a long time trying to understand why that happens, so persistently, so predictably, especially in otherwise very intelligent, lucid and principled people. I knew it had to be something vaguely about self-deception. Herbert Fingarette’s meditations are helping, gradually.
Thanks for this. It exposes the game in all its corrupt splendour.
Report comment
*lumateperone was not being tested in “people with schizophrenia
Maybe all people with real schizophrenia are already gone and the doctors just want to fix their mistake and use the pills to create new ones, from my point of view this is quite likely. How to count people who supposedly heard quiet music without a radio. It remains only to eat garlic and engage in psychotherapy.
Report comment
Thanks Robert.
I never read to the end.
I felt sick, not only by already knowing that there is no safe drug and already knowing that EVEN if there WERE a drug developed with less ‘side effects’, it would just be a drug with possibly even worse long term effects. Obviously it is taking old recipes and tweaking them, and giving them a new name.
What made me feel really sick was the experiments that resemble no less than something worse than bloodletting.
Experiments on people. Really just treating them no better than rats.
So a person who was on these drugs long enough to conveniently be affected enough to not realize what they are committing to?
When will we have involuntary trials? I am thinking these were in a wretched manner. So how do we and should we test people for ability to knowingly consent?
Are these patients ‘allowed’ to make wills? would they be ‘allowed’ to make decisions of life or death?
Sick. making people withdraw in an “inpatient setting”, so no one can watch. This is where we need a watchdog. To protect patients. WHO was there watching? Family? And if not, WHY NOT?
Are people with these labels “vulnerable”? Then why allow them to be ONLY with those that medicate?
Report comment
To induce a proper sense of Stockholm Syndrome. Uh–insight I mean.
Report comment
So Jeffrey Lieberman is lauded as he makes himself and other rich people richer through spinning fraudulent research based on outdated theories, giving the medical industry and the government excuses to prolong the harm and profits from neuroleptics. (Even as we read inflammation is emerging as the promising new frontiers of brain and body health [https://www.theguardian.com/commentisfree/2020/jan/19/inflammation-depression-mind-body]). It looks like our governments will continue to waste our taxes to pay more for the latest and most expensive neuroleptics, however damaging and dangerous these drugs, and will continue to force them into our bodies as they manage our behaviors in schools, prisons, child and elder care, military fields, hospitals and in the street. So strange to consider corporate crimes against humanity being rewarded with millions while good citizens in the Southwest are putting water and food out for people being forced to migrate (often thanks to our own oppressive policies) and charged with criminal offenses which can lead to years of incarceration for helping others (see https://www.madinamerica.com/2020/01/interview-liberation-psychologist-mary-watkins/). However terrible and topsy-turvy, I appreciate being able to learn what’s really happening in this world we care about whenever I visit MIA. May we be emboldened by what we are learning together and act on it as we can. Thank you again Bob.
Report comment
Lieberman makes himself still richer by turning even more people into penniless cripples. Who slowly die.
People like him deserve the reward Farid Fata got.
Report comment
Thanks so much for forensically shining a spotlight on this Robert. My son took himself off antipsychotics (over 8 months) when he realised they weren’t doing anything beneficial. I’m seeing kids strongarmed onto these who aren’t even psychotic and they are just meekly accepting this is their fate, assuming the drugs have been trialled on kids like them. As we all know now, thanks to you, they haven’t. Thank you.
Report comment
Most people don’t have “mental illness” to begin with
– they either have “substance problems” or “psychological problems” (hallucinations and delusions included).
The only Factual Recovery from “Schizophrenia” on record, comes from people that reject Psychiatic drugs in favour of a Non Psychiatric approach (often of the ‘Spiritual’ Variety).
Report comment
Dear Robert
On the Subject of The Unanswered Question:
Maybe the psychiatric experts were so used to interpreting or misinterpreting symptoms of normal behaviour or so called mental illness at their own convenience – that they had lost the ability to tell the difference (I’m serious).
It’s not only in Psychiatry:
https://www.motherjones.com/kevin-drum/2020/01/health-care-is-an-8000-tax-on-every-american-family/
“….Despite paying $8,000 more a year than anyone else, American families do not have better health outcomes, the economists argue. Life expectancy in the United States is lower than in Europe…”
Professors Angus Deaton and Anne Case.
Report comment
Thank you Robert for your ongoing vigilance to investigate and report on drug studies that are clearly skewed, or as you put it well in Anatomy of an Epidemic “biased by design”. (an incredibly informative book) The JAMA article states this drug approval is “potentially exciting” – yes exciting for those who stand to make big profits from having a new drug with a patent as opposed to older generic drugs. Only those deemed to respond well were selected and – 70% of the people in the study were prescribed lorazepam – when adding another drug that combats agitation, anxiety and symptoms of debilitating EPS certainly skews the results. Everything about this study smells bigtime. Thank you for this excellent investigative report, and the MIA website and all you do to educate people and help save lives.
Report comment
There’s one interesting data-point, the half-life of Lumateperone (similar to clozapine) is advertised at being 13 hours. Olanzapine is almost 3 times that (33 to 51 in the elderly). It would account for the less side-effects reported. It get’s out of the system faster. (Unless you give it multiple times per 24 hours).
Report comment
Thanks Robert Whitaker for this article on lumateperone. The only thing impressive about the drug was the phenomenal rise in stock price. No one will be selling me on this “new” drug no matter how bad things get.
In my 50’s I quit anti-psych drugs. That was 14 years ago bc they didn’t work. Over months of increased dosage & different drugs nothing improved. I was still working & in fact the drugs effected my job performance & made everything worse. After low dosing on one very “new” anti-psych I had to crawl up the stairs of my walk-up on all fours.
One voluntary trip to a hospital ward made everything worse. I’d never used street drugs, didn’t abuse alcohol & still don’t. Never had thoughts of suicide until after entering the ward of a NYC hospital that happened to be in financial straights & about to close. A big mistake.
The back story of the Soderbergh movie, Unsane was likely applicable to what happened to me inside. That movie took on corporate hospitals & short term ‘stressed out’ rehab that then gets magically turned into long term with good insurance being the determining factor. This is extreme untouchable territory in society generally among the left in particular. A great movie regardless of reviewer fixation on camerawork.
I mentioned my hospital stay to my GP. Another big mistake. His overdone reactions were meant to be seen as almost comical. He did the “I’m not a psychiatrist” routine etc.
Report comment
Big Pharma has to deluge the market with dubious antipsychotic drugs. NAD (nicotinamide adenine dinucleotide) is coming on the market. It has the same antipsychotic properties as “straight” niacin, but works much faster in the treatment of various “schizophrenias”, which must be hidden to preserve the market for allegedly antipsychotic drugs, as it’s unpatentable.
Report comment
Lumateperone was a guaranteed money maker due to the psychiatric guidelines that are laid down by the APA which is that the newest drug gets used first in practice even though practitioners have much more experience with older drugs and the fact that anti-psychotics are largely funded by medicaid dollars which are tax payer money whether it is $900/ month or $1,200/month. The guideline itself guarantees instant market share to any new drug “discovered” and approved in the effort to “treat” the severely mentally ill.
This was a win for everyone except the patients and the public. The patient does NOT matter anymore and outcomes don’t matter either.
Report comment
….thank GOD for Mr. Robert Whitaker! Robert, your article is “TLDR” for me, but it’s not your EXCELLENT writing and analysis. I have a very low tolerance these days, for the psychobabble and pseudo-medicalized gobbledygook of PrRMA and the slimy “Dr” LIEberman…. at least he’s aptly named….
The salient point is the one-day rise in stock valuation of over $1Billion. It’s all about MONEY, and POWER, and CONTROL. If this drug is so good, why isn’t it only available as a non-profit public good, instead of the PRIVATE, FOR-PROFIT PRODUCT which it ONLY is available as? I’d love to hear “Dr.” LIEberman’s answer to THAT question! But it would be a lie, so, who cares….????….
Report comment
So true Bradford,
Why not hand out these drugs for free? And also good housing?
Obviously we should take care of our people.
Especially the MI ones. They should not be punished by getting substandard housing or lack of care.
In fact, we should give extra, things like meals in a restaurant should not be a luxury. Things like shopping for good sheets and pillows or having money to take someone on a date, attend a play, these should be available especially to those who are told they have a hopeless disease.
The least we can do is provide adequately for the ‘diseases’ that the sufferers are not responsible for.
Psychiatry somehow makes sure the diagnosed just have a shitty life til the end.
Report comment
Shrinks express sorrow at the “stigma” their test subjects experience in being denied housing, jobs they could perform despite being drugged up and having family and friends reject them.
Yet they refuse to accept any responsibility for the situation and love to go on national TV to tell the world how dangerous/stupid/evil all their “patients” are. As Paula Capplan points out if I tell someone you have tuberculosis and they avoid you does it make sense for me to express outrage over how they treat you?
Report comment
” As Paula Capplan points out if I tell someone you have tuberculosis and they avoid you does it make sense for me to express outrage over how they treat you?”
Bears repeating.
Report comment
And – very importantly – this new drug can also cause tardive dyskinesia. The prescribing information leaflet even states after mentioning TD: “In patients who do require chronic treatment, use the lowest dose and the shortest duration of treatment producing a satisfactory clinical response. Periodically reassess the need for continued treatment.” (page 2 here: http//www.intracellulartherapies.com/docs/caplyta_pi.pdf).
But we all know that people with a diagnosis of schizophrenia are usually put on neuroleptics for life… I came off a neuroleptic about a month after my first and only psychotic episode in 2012 (I was diagnosed with “schizophrenia” after this episode). Coming off the neuroleptic has been one of the best decisions in my life. I have never had a psychotic episode again despite my diagnosis…
Report comment
Joanna
I eventually managed to come off my ‘long acting neuroleptic injection’ many, many years ago through a very careful drug taper, and psychological means – similar to below:-
https://youtu.be/3I5L2otW4r8
I’ve remained well since, so there is no such thing as “Schizophrenia”.
Report comment
Fiachra, great to know that you, too, came off neuroleptics a long time ago – and I fully agree with you that “schizophrenia” does not exist! I have recently noticed that some young men who have been using street drugs are diagnosed with “schizophrenia” in my country. Sadly, many of them think that they really suffer from a severe mental illness and become long-term psychiatric patients…
I have recently had to tell my own brother who had spent some time on a locked ward because of suicidal thoughts that he, too, might one day be diagnosed with “schizophrenia” if he is not careful…
Report comment
The extension of “schizophrenia” is amazing and frightening.
Report comment
Let me just chime in to remind everyone that there is no such thing as “schizophrenia.” It is a figment of the psychiatric imagination.
Report comment
Glad someone said it!
Report comment
No, but there are a number of conditions which can mimic the syndrome shrinks mistakenly call schizophrenia. As far as I know, psych drugs have positive effects on few to none of these mimics, other than to perhaps give you dyskinesia, should you be daft enough to call that a positive effect.
Report comment
Thanks Slayer for the reminder that the label is an opinion.
Report comment
Thank you.
I would say that it’s worse than an opinion. So-called “schizophrenia” is a figment of the psychiatric imagination. It is the sacred symbol of psychiatry, and psychiatry is a pseudo-scientific system of slavery that masquerades as a medical profession. Brain damaging drugs are not a cure for fictitious diseases. In fact, brain disabling, psychotropic chemicals CAUSE the very damage that they purport to remedy.
This is just one of the myriad reasons why psychiatry must be abolished.
Report comment
No, “Slaying….”, I’m really not disagreeing with you here, in any fundamental way. But yeah, I want to challenge the characterization in language. I’ve come to see that ALL so-called “mental illness”, including of course, “schizophrenia”, is in fact the result of trauma, and/or abuse throughout one’s life. This would include toxic chemical exposure in the womb, and Mother’s lifestyle. Many of the influences are very subtle, but theyu ALL add up, and in unique ways. But again, trauma & abuse, abuse and trauma. The sequelae of abuse and trauma is studied ever more closely. Currently, the acronym “ACE”, for “Adverse Childhood Experiences” is in fashion, and it’s big step forward. Abuse and trauma does come in something like patterns, and we can see certain patterns in persons, also. So in some persons, this all adds up to an extreme degree compared to most folks, and it’s these folks who get labeled with tags such as “schizophrenia”. So there really is a “there” there, but why call “it” “schizophrenia”? Easy answer. Money, power, and control. The original “schism”, or split from the society of other people, leads to the belief that so-called “schizophrenia” really is a real thing. And if we call an arbitrary label a “diagnosis”, then we can use that alleged diagnosis as the basis to $ELL DRUG$, and MAKE MONEY. So, MONEY, POWER, and CONTROL. For the already rich, powerful, and in control. So I can’t agree that “there’s no such thing as schizophrenia”, only that “schizophrenia” is an arbitrary, wrong, and outdated concept. I say it’s just as bogus as you say, but I still say there’s a “there” there, but that “there” is NOT best described as “schizophrenia”. What do you think about what I’ve said here?….
Report comment
Exactly, anomie. Exactly.
Whitaker and others do excellent work on the very surface of psychiatric fraud. This is helpful because people who read can begin to question the psychiatric paradigm. But rarely do people have the courage and the foresight to dig deeper into the myth of mental illness. Karl Kraus and Thomas Szasz had greater courage and understanding than anyone writing today, but their works have largely been ignored, ridiculed, or forgotten. That is often the fate of truth-tellers.
Report comment
🙂 #shalom
Report comment
Or pretended to treat non-existent heart diseases and prescribed pills to damage the hearts of your patients. Then sold more drugs to treat the damage you caused which caused more damage in other vital organs.
Now all that’s needed is more effective marketing for replacing those pesky patients who keep dying.
Report comment
I used to think Psychiatry couldn’t be categorized in up down, wonderful or toxic terms. I still don’t think in either of those extremes. But given the choice of sounding like a fear & doom, all is lost type so common everywhere these days & defending P-docs, I lean towards seeing psychiatry as my P-doc described in 2007 when committing the sin of truth regarding anti-psych prescription drugs, “we’re still in the dark ages.” This was before prescribing Provigil for someone with insomnia. Which drove me into sleep deprived delusions even after I stopped taking it.
Report comment
Thank you Robert for this analysis.
I would add the drugs sexual effects on the brains D2 receptors has not been reported/studied. Numerous rat brain studies show haloperidol and clozapine stop male sexual behaviour.
Modulation of Dopaminergic Pathways to Treat Erectile Dysfunction.
https://www.ncbi.nlm.nih.gov/pubmed/27541930
switched to
Modulation of Dopaminergic Pathways to Create Erectile Dysfunction.
Report comment
To better summarize what I just wrote, I’m skeptical of all sides of the argument, mainstream psychiatry, alternatives & indictments of the existence of certain entries in the DSM & those applying it. Which also means I might find things that are useful in each of the categories mentioned.
Report comment
Truth is more useful than illusory neutrality.
Report comment
Yup, everybody thinks they know what “the truth” is. I’m not neutral I’m skeptical and stay away from the lingo of the moment.
Report comment
If you had Akathisia you would know the importance of:
“Never get out of the boat”
https://www.youtube.com/watch?v=_4dFDBYWuTc
Here’s the current problem:
The tiger is about to leap into the boat.
Report comment
“Yup, everybody thinks they know what “the truth” is” – I hope that you don’t exclude yourself from this categorization.
I’m skeptical of your skepticism. 🙂 But I agree that we need to dig deeper than what you call the lingo of the moment. That’s why I recommend Karl Kraus and Thomas Szasz. They weren’t right about everything, but they were truth-speakers long before anyone here was born.
Report comment
I was a “true believer” in Szasz long ago. It kinda gets old. I recommend you read “The Inflamed Mind” by Edward Bullmore (yup, Bullworth-like Brit name and all).
I spent the last 10 yrs watching different platforms and comment sections get swamped by “all is lost” angry end-timer burn-it-down anarcho types. Here it’s toxic this, criminal psychiatry that, SCZ doesn’t exist etc. IOW, the same stuff I was reading in 2007.
The best that mainstream can do is what they were doing 15 yrs ago, going after stigma bc they haven’t got anything better to say. It’s a good thing to go after stigma, very safe. Everybody gets the feels. Both extremes repeating repeating and subconsciously waving to each other.
Report comment
Mainstream psychiatry hearts stigma.
Report comment
Szasz never gets old, because he tells the truth about psychiatry and the need for abolition. “The Inflamed Mind,” on the other hand, relies on the fiction of so-called “mental disorders.”
Report comment
Try and think of schizophrenia as a collective noun for unknown problems in human thinking, behaviour, experiencing, perceiving and interacting.
Also, dragons aren’t real.
Report comment
I am glad society sees it that way.
What collective noun do we use for drugs?
What collective absolute for distinguishing illness from non illness?>
What do we call it when a collective noun causes abuses, control, lack of medical care, no rights left.
We have as to date no studies done on shrinks that when removed from their jobs as shrinks, say over a ten year study of all shrinks (with no money in their accounts)
How they function. What jobs they choose.
Let us all be okay with psychiatry and the abuse, lies.
But alongside of “schizophrenia”, let us list the lack of medical care that non criminals get, the clout and validity in court that non criminals receive.
Let us be truthful that it is now a UN issue. To have an illness that presents “problems in thinking”, the onus to prove that it presents an inability to think logically or act logically in every single area, that onus is on who? How do we prove that?
And so, if it is down to the word of a shrink, it is very obviously not proof.
I see Britney Spears has few rights, or no rights, but she does not have schizophrenia.
I also wonder why every man and his dog uses these words of illnesses to prove MI exists? It is the public bait word. “Anxiety” seems so average.
Report comment
₽ or $?
Report comment
You can try that if you like, but that’s not what so-called “schizophrenia” is. That’s not how it was invented, and that’s not how it operates in the real world.
https://psychiatricsurvivors.wordpress.com/2016/06/19/schizophrenia-the-sacred-symbol-of-psychiatry/
Report comment
There is no such thing as a “real world”.
Report comment
Removed for moderation.
Report comment
???
Report comment
sam plover asked, “What is the collective noun for drugs?”
That’s a tricky one because drugs is the collective noun for more than one kind of drug. Or drugs could be the plural for a singular noun.
Report comment
sam plover wrote “What collective absolute for distinguishing illness from non illness?>
What do we call it when a collective noun causes abuses, control, lack of medical care, no rights left.
We have as to date no studies done on shrinks that when removed from their jobs as shrinks, say over a ten year study of all shrinks (with no money in their accounts)
How they function. What jobs they choose.
Let us all be okay with psychiatry and the abuse, lies.
But alongside of “schizophrenia”, let us list the lack of medical care that non criminals get, the clout and validity in court that non criminals receive.
Let us be truthful that it is now a UN issue. To have an illness that presents “problems in thinking”, the onus to prove that it presents an inability to think logically or act logically in every single area, that onus is on who? How do we prove that?
And so, if it is down to the word of a shrink, it is very obviously not proof.
I see Britney Spears has few rights, or no rights, but she does not have schizophrenia.
I also wonder why every man and his dog uses these words of illnesses to prove MI exists? It is the public bait word. “Anxiety” seems so average.”
I believe that the study of language is worthwhile. And that such a study would settle a lot of these questions that you’re asking.
Reading you I *feel* as best I can your anger about these issues concerning words and language and how words can oppress and abuse and hurt and trap.
In terms of proving sanity or madness that’s only ever going to be a social and cultural set of problems, arbitrary decisions, and paradoxes.
One way out of that is to accept that we are all of us broken, individually and collectively. And that humans are fundamentally self-deceived and self-deluded. Almost all that we hold as true and constant and reliable is anything but… however, we hold fast to our many delusions and deceptions, and if we didn’t, it’d be the worst nightmare you could ever imagine.
I;ll close by confessing that I have no emotional or intellectual interest in Britney Spears. Every time her name is mentioned an infamous paparazzi image of her comes to mind and it wouldn’t be right to go about describing it but that ultimately is what she is to me. Although of course she is a human being, and I hope she has people in her life that love her and protect her and remind her to check herself before climbing out of the back seat of a limo.
Report comment
Uneasy rests the head that wears the crown.
Report comment
Geronimo,
Not sure about the book you mention,
It seems to be written by someone who worked for glaxal
and was pretty much defending psych meds.
Report comment
The author is definitely an insider Cambridge guy. A Neuropsychiatrist with a PhD in philosophy. He’s a skeptic who (way too low key for many on this site) takes his profession and prescriptions (depression, anti-psychs) to task. He says anti-inflammatory drugs *as a starting point* might be a whole lot better for depression than what’s available now. (No kidding) He goes on about the conservative nature, frustration and slow to no progress of his profession.
How long have I been following the whole brain inflammation argument? Its nothing new for me both in functional physical medicine and glial cell brain immunity. Depression prescriptions for me were a brain fog joke. Same with the 3 days I was on Benzos and they actually worked for anxiety. That said, I’m not in the, ‘those Glaxo guys are all in on it’ bubble.
Report comment
I read an article in the UK Guardian by Edward Bullmore. Glad he’s taking his colleagues to task.
Psychiatry’s standard treatments cause inflammation. If there’s any validity to Bullmore’s hypothesis, what his colleagues are doing is exacerbating the original complaint.
Report comment
Looks like some people are bailing on the shares.
Report comment
Rachel777, I also read that recent article. This inflammation idea has been floating around mainstream for 10+ years. That there’s less of a divide between brain and body. In traditional oriental medicine it was assumed. I was reading the book when I read the article and I’m still reading the book. It’s not filled with hair-on-fire indictment exclamation points and he’s not breaking any new ground. Which means he’s not going on about stigma bc there’s nothing else to say and he’s not in the “all is lost” burning down his own professional house.
Why so slow? Bc there might not be any big breakthrough new exciting drug to cause exponential increases in some stock price. That’s the easy explanation. Anyway the wheels have been turning long enough to force improvement. I’m just as tired of hearing the inflammation argument as I am the ‘lock up the P-docs’ and force them to take their own prescriptions scripted lines.
Report comment
“Scripted lines”? Nobody writes my comments but me.
Just a fantasy for any of us. Of course. Like getting back the lives they ruined.
Preventing them from locking up, crippling, and killing more people would be enough for me.
My body is inflamed and my brain damaged from the treatments. My heart was going bad before I escaped. Psychiatry was killing me. As it has countless others. My “bipolar 2” was created by a drug reaction no one acknowledges.
What’s left to improve about psychiatry? They get 6 or 7 digit salaries and never have to worry about lawsuits. The Professional House is in great shape. No one is about to burn it down. Like the pigs at the end of Animal Farm. They get what they want and that’s all that matters.
Lots of people come here to mock the suffering. “Psychiatry is not going anywhere!”
My response? Doing right is more important than winning.
It’s better to lose fighting for what’s right than win by joining the forces of evil. If Hitler had won World War II do you think opposing him would still have been worthwhile? (It cost Dietrich Bonhoeffer his life.) I say Yes.
Report comment
The planners of WW2 never intended Hitler to win, his only role in the puppet show was to lead Germany in fighting WW2, NOT “win” it….
BOTH WWpart1, and WWpart2, were carefully pre-scripted by the Billionaire Boys Club (BBC)….
Report comment
I note the shift in narrative regarding Ukraine in our media in Australia. The people who were encouraging the war with Russia now making it sound like they were against it in the first instance. I guess they must be running out of money for the weapons these people were selling them?
Report comment
Yes, the GRIFT has been fully exposed….it’s a fake war, a centuries-old border clash pimped up as a serious war. Massive money-laundering propping up the whole murderous charade….Plus, a live-action field test for new digital & drone technology & tactics….the ARMS Industry loves it! And gets fat cat rich off it, the longer it drags on….
Report comment
I also read that recent article. This inflammation idea has been floating around mainstream for 10+ years. That there’s less of a divide between brain and body. In traditional oriental medicine it was assumed. I was reading the book when I read the article and I’m still reading the book. It’s not filled with hair-on-fire indictment exclamation points and he’s not breaking any new ground. Which means he’s not going on about stigma bc there’s nothing else to say and he’s not in the “all is lost” burning down his own professional house.
Why so slow? Bc there might not be any big breakthrough new exciting drug to cause exponential increases in some stock price. That’s the easy explanation. Anyway the wheels have been turning long enough to force improvement. I’m just as tired of hearing the inflammation argument as I am the ‘lock up the P-docs’ and force them to take their own prescriptions scripted lines.
Report comment
The problem resides in trying to come up for a “solution” to something that can not be identified or defined as a single entity. “Mental illness” is a concept, not a disease state with a defined cause. Why would we imagine for a moment that something so nebulous and subjective as “mental illness” could possibly have a unitary cause or solution?
Report comment
The problem with that is thinking there’s no physical correlation to mental illness. Which immediately summons more scripted counter arguments around for decades about missing chemical imbalances. It’s been pretty much accepted in theory that SCZ is several conditions combined. Among those are inflammatory processes as cause not just effect.
I stopped anti-psychs long ago. I recently found a mote of dust under a heavy piece of furniture. I am sensitive to dust and have been breathing it in for years. I’ve always had asthma so when I was short of breath 4 yrs ago on my way to the gym I used my inhaler but then also stopped the gym. I took a calorie loaded coffee drink with collagen bc I liked the taste. Anti-inflammatory effects were a distant second. A month later I stopped needing the inhaler. It wasn’t psychosomatic. I made the mistake of using the drink with a meal when it was supposed to be a meal substitute. I was not exercising while breathing in all that dust and suppressing breathing symptoms which meant all kinds of accreted crap accumulating internally in reaction.
Long run-on story short, multiple factors get lost while thinking that I had relieved asthma (no, I didn’t think I’d found the cure) which is an inflammatory condition while along the way making things worse. (Lungs made of different kinds of collagen proteins vs collagen protein gets digested so therefore couldn’t be a factor etc) 3 years later vitamin shelves are loaded down with all kinds of keto collagen powders some of which might actually work. Which means there’s something to it and also means the cheaper stuff has impurities etc.
All of that involving a physical condition and the fact that I didn’t like taking a prescription spray 2 or 3x a week. Mental factors that I still contend with have correlations likely much more complicated. But still solvable.
Report comment
There is nothing in my statements that conflicts with the idea that certain people’s conditions might have a physiological/medical genesis. All I’m saying is that calling these “mental illnesses” obscures the fact that there are multiple possible causes and multiple possible solutions, and that in many cases, there need not be anything physiologically wrong at all. Once we say someone “has depression,” any attempt to understand the real causes, be they physical, psychological, spiritual or social in nature, come to a rapid end. This is particularly true when the system automatically assumes that every single possible divergence from complete satisfaction with the status quo is caused by a “chemical imbalance” or other physiological problem.
There are most definitely physical illnesses that manifest with psychological “symptoms.” It just doesn’t work the other way. As Socrates pointed out, if all men have facial hair, and Bob has facial hair, it doesn’t follow that Bob is a man. The logic flows from known physiological conditions to psychological effects, but not the other way.
Report comment
The real problem is not that the inflammation theory is totally bogus. The real problem is that when there IS a physical illness contributing to someone’s mental suffering, the psychiatrists have ZERO interest in finding it. I continue to recover from Lyme Disease. I am a thousand times better than I was a year ago after having had a month of antibiotic treatment. I mourn for the time lost to psychiatry when what I mostly needed was an understanding empathetic ear (for my traumas) and a long course of antibiotics for Lyme Disease. Psychiatry greatly hindered my ability to recover by providing brain damaging “treatments” and shrugging it’s shoulders as I slowly deteriorated.
Report comment
I don’t think any curious theory is bogus, but I also can’t accept it as truth. So I guess it is bogus until proven to be correct.
I also have no interest in harmful drugs created by theory.
Look what we are swallowing because of theory.
And the other thing is, MI, it will still be looked at the same way, socially and politically. It will still be the DSM, still be forced drugs, still little kids being drugged.
I do believe that psychiatry invents “possibilities” for one major reason. To keep the MI idea going, in those who started to doubt.
Shrinks are catching on, that their drugs are being proved as bogus. So let us invent new theories which will awaken hope in the doubters. The on the fence people like the sound of new possibilities and then, we are right back to the same old.
The DSM and it’s powers needs to go, they can still tinker away with inflammation and a few of the other theories.
Besides, new possibilities sells a few books and greases the machine, revs up big pharm. OHHH, so many possibilities.
Report comment
Geronimo,
So now it’s inflammation?
Then ALL of psychiatry has to back out and say, oops we were wrong.
And the same psychiatry then creates drugs for “inflammation”….but this time they are right?
So this inflammation is the cause of suffering?
No entertaining the idea that you and I simply exist because of genetic variations?
That as a society we can’t all be shrinks? Some of us must be MI for shrinks to exist?
We are after all, the closest to a virus
Report comment
“So now it’s inflammation?” Where have you been? Nothing new about it. Functional MD’s have been going on about it for many years. I mentioned previously the “all is lost” “end-times all the time” pervasiveness I saw swamping YouTube and the Internet around 2007. That *nasty* book by the Glaxo guy I mentioned pushes hard against closely held BBB dogma within medicine and psychiatry. It also pushes against dogma closely held on this site. Would I see a P-doc again? A functional neurologist? Or figure it out myself?
Just like last year I’m probably still recovering from the flu shot I got in Dec. How is that possible? Am I now anti-Vaxx? No. My previous comment might explain it. I’m probably still recovering from breathing in crap for years and the jab makes it worse for awhile. Yup, my fault. Not the flu shot, not the P-doc, not the bogeyman or God(ot)etc. Still in Room 101. 1984 wasn’t just a cautionary about future dictators. I don’t listen to the cognitive dictatorship Big Brother pessimism in or outside my head. Sometimes that ain’t so easy.
Report comment
https://youtu.be/zz4YVJ4aRfg
Something different from 2016 having nothing to do with drugs or P-docs from Room 101. Saw this video for the 1st time, saw her before. Another too good to be true non-miracle non-drug I first heard about last year.
Then there’s this per Wiki: “Although there has been some basic research on how sulforaphane might exert beneficial effects in vivo, there is no high-quality evidence to date for its efficacy against human diseases.” Her background is biomedical research and she basically quotes the research of others. I can almost anticipate, ‘What? Sprouts? You use Wiki? Where’s your proof deuter’
I don’t dismiss it, haven’t tried it, I will try sulforaphane in the form of sprouts and powder after listening to her on different venues for the 5th time. She’s good to look at also. 28:00 minutes in she starts talking about, guess what? Inflammation, SCZ, depression. 38:00 Dr Patrick does a recap.
Report comment
Latest Alzheimers research on rats. Crossing the old BBB with very low dose lithium stops early stage. Positive results from low dose lithium for other conditions is also not new.
“From a practical point of view our findings show that microdoses of lithium in formulations such as the one we used, which facilitates passage to the brain through the brain-blood barrier while minimizing levels of lithium in the blood, sparing individuals from adverse effects, should find immediate therapeutic applications,” says Dr. Cuello. “While it is unlikely that any medication will revert the irreversible brain damage at the clinical stages of Alzheimer’s it is very likely that a treatment with microdoses of encapsulated lithium should have tangible beneficial effects at early, preclinical stages of the disease.”
Report comment
@Geronimo
Not just spouts. Cabbage, broccoli and turnips too. Just don’t boil them. To do so is to torture and kill all the goodies they offer. Steaming is best.
Lifestyle choices are obviously going to be good for the body, the brain, general wellbeing. A lot of these excellent choices then get knocked out by polluted air and alcohol, but best try and run on a full tank in a polluted, dirty world, I reckon.
I wonder what effects all the plastic particulates are having on our bodies and brains? The brassicas will be full of them.
Thanks for the link. She talks too fast for me. But Youtube allows you to slow down or speed people up in the settings.
Report comment
Just had my 1st batch of washed broccoli sprouts with just salad dressing. Not bad not good. It needs the dressing. Will supplement with the powder when I get it.
I try not to think about horrendous forever chemicals but I don’t cook on or ignore them. I have a constant thirst which seems to go with big time insomnia, alcohol & the MI territory in general. Stayed away from alcohol so far in January.
Dr Patrick reminds me all too much of some1 I met up with last year. Must be my disordered or wishful thinking. Except there was nothing disordered ab’t it then or now. Famous last words. You should catch her latest on NAD. She says the jury is still out. Been trying to find the Dave Asprey link to NAD supplements I read about 6 months ago.
Edward Bullmore (great name) is in the news again with a quote on fMRI developing adolescent brain scan models. That would raise all kinds of hackles on this site. Still reading his damn book. Insomnia screws up my reading habits.
Report comment
Hi Everyone! While we all agree psychiatrists have the power to destroy their patients in the name of care and are responsible for what can only be described as ‘The Silent Holocaust’, it is my experience and with my somewhat limited knowlege of history that what goes around comes around. The people are in fact only human and if they look after themselves in their elite lifestyles then they will inevitably grow old and find themselves in your caring comfortable nursing home. Where lets face they might be susceptible to other caring professionals who just might see fit to prescribing the odd anti-psychotic or anti-depressant to the old dears. In fact it is my experience that ‘What Goes Around Comes Around!’.
I draw cartoons and hopefully this will come through to you and it is one of my favorites.
https://mail.google.com/mail/u/0/?tab=wm&ogbl#inbox/QgrcJHsbcVPCvXtbrqtMSxNpzblvSqJXwtV
Report comment
Might it be that the goes around, comes around ends up with the political masters who have enabled these human rights abuses might become victims of them also?
Old Adolph was riding the crest of a wave there for a while (along with his cronies), but took quite a fall once he was addicted to the drugs he was being prescribed by the same people who put his ‘delousing’ program in place with his enabling laws. It’s always a shame to see the effects of the sins of the father being punished but it may be their children being subjected to forced ECTs for a single facebook post misinterpreted by their teacher.
I’m a bit like the doctor who interupted my unintended negative outcome in the Emergency Dept, I don’t have the stomach for it. But I know some folk who do.
Thanks for reminding me of the potential outcome of this “Silent Holocaust” Bippyone
Report comment
“there has never been a ‘randomized, double-blind, placebo-controlled study’ of an antipsychotic in medication-naïve patients”
Of course there has: real life. The millions of successful first episode patients treated with standard medications. The half who do not respond adequately, if at all, to standard antipsychotic medications logically have different brain diseases that present similar symptoms. Psychiatry’s failures are almost the exact opposite of what MIA constantly claims. The issue is one of improper scientific categorization, which MIA shares with the worst elements of the Psychiatric establishment. What’s needed — and families of victims could certainly do this even without professional input — is to acknowledge that, 1) Schizophrenia is a syndrome — not a disease — describing specific symptoms of brain dysfunction, and 2) that we can logically identify ever-more specific brain diseases (and other explanations of the characteristic symptoms) primarily based on the efficacy or lack thereof of the full spectrum of medications available.
When it comes to scientific trials, the standard that you hold any possible brain science to is a brute impossibility. Real life has to be the judge of effectiveness, or there can be no progress at all. It would be profoundly unethical to risk the lives of patients, most of whom have sustained brain damage such that they lack the ability to comprehend that they have been injured, most of whom are homeless or without family or advocates, by changing previously successful treatments. So, you are already filtering out the worst afflicted victims who couldn’t possibly participate. So, they have to look for hints among ideal candidates, then release the molecule to the wild and hope that it eventually reaches the most vulnerable among us and miraculously works. That’s how medicine has to work.
Lastly, if you turned your critical eye toward an analysis of anti-materialist forces of stagnation and prolonged suffering and violation of the right to progressive functionality, there is zero scientific evidence that trauma causes cognitive disability. There is plentiful conflation of effect and cause and counterfactual speculation, but zero scientific evidence linking a negative psychological event to a change in brain function. And MIA regularly filters out the negative stories about untreated psychosis. Is it more likely brain damage is far more prevalent than the establishment would ever want to imagine paying for? And that “trauma” and the sum of psychological explanations for obvious physical disorders is the greatest superstition ever perpetrated on the seriously ill and their families? Of course and of course. Again, it’s real, material life that is the final word on why over 70 million human beings are allowed to suffer from severe cognitive problems and what solutions actually give people their lives back. Establishment Psychiatry and MIA are two sides of the same coin, chronically oversimplifying categories defining the problems and overcomplicating attempts to find solutions.
Lumateperone is a progressive step for an industry trying to eliminate negative side-effects, meaning they are acknowledging that what they offer isn’t good enough, and Psychiatry shouldn’t be content being vague. It cannot be judged until it’s tried by real people in the real world, and unreasonable criticism shouldn’t be used to manipulate victims to create a bias against it right out of the gate. The only result of the sentiment of this article is to enshrine older medications definitely known to have serious harmful side-effects for another 70 years, or to keep pretending there aren’t knowable subcategories that explain the common symptoms of psychosis, which causes victims and families to do nothing or give up. Absurd and unethical.
Report comment
Thanks for your passionate reply.
There are a couple of things you say that I have to take issue with. First, there is plenty of evidence of significant brain changes due to trauma. This was the primary finding of the “Decade of the Brain” research, which was actually quite different than what was anticipated. More importantly, and less well known, are the findings that the brain can continue to change in a POSITIVE direction when a traumatized person is supported by a healthy adult caretaker or support system. Dr. Bruce Perry is one of the best resources for this. Easy internet searches for these items. Here is one just to get you started: https://www.psychologytoday.com/us/blog/the-mindful-self-express/201809/how-ptsd-and-trauma-affect-your-brain-functioning
You also seem to conflate “cognitive disability” with “mental illness.” You suggest that most of those severely affected by the syndrome called “schizophrenia” are affected by brain damage. I know of no evidence that this would be true in most cases – there has been a long, intense and frankly biased search for brain damage associations with “schizophrenia” and many other “mental disorders” with little to no results. There is evidence of brain matter loss in people diagnosed with “schizophrenia” over the long term, but Nancy Andreasen’s own research, which she really didn’t WANT to believe at first, showed that antipsychotic drugs cause loss of brain matter when used over the long term, so such studies are meaningless unless controlling for AP use, which is almost never done.
Finally, you also assume that the “schizophrenia syndrome” is due to different types of “brain malfunction.” As I’m sure you are aware, science proceeds from hypothesis to proof, and it doesn’t work to assume the conclusion that brain malfunction is the cause when that has not been shown to be the case. And your suggestion that the fact that certain drugs “work” to decrease these loosely defined groups of “symptoms” proves an underlying disorder is similarly flawed. Under that reasoning, alcohol must be addressing an underlying “brain malfunction” because anxious people feel better when they get mildly intoxicated. Correlation can’t be used to diagnose anything.
I very much respect your reframing of “schizophrenia” as a syndrome, something which would really help if we all operated from that assumption. Unfortunately, the current reality in “mental health” research is that these syndromes are being treated as unitary entities for investigation, and that leads to a lot of misleading, trivial and/or meaningless results. I also agree that it is easy to “throw the baby out with the bathwater” if one is too committed to dogma on either side of these questions. I’m a scientist in the end, and at this point, I see no science that really proves that “schizophrenia” is caused by any kind of a brain malfunction, and I am doubtful that continued research will prove any such thing. Partly because, as you say, it is a syndrome, and there may be (probably are) some subgroups that DO have biological causation, but these will never be discovered as long as research is done on “schizophrenia” with the assumption that all cases have the same cause and that this cause is necessarily biological in nature. It is clear from research that trauma has a very high correlation with the syndrome in question, as does urbanization and migration to a foreign culture. It’s also very clear that the manifestations of this “disorder” vary widely depending on the culture in which they occur and how the culture in question responds to such issues. It is, as you suggest, a much more nuanced picture, and until proven otherwise, I think it makes a lot of sense to assume that both nature AND nurture are involved.
Report comment
“first episode”…..We have no way to duplicate such events, as there are MANY people that will suffer one episode and never again, even without drugs.
There are many who might “suffer” a few episodes throughout their lives.
Should all these people take chemicals, just in case?
Report comment
Of course its possible to have an RCT in FEP, McGorry just did it with small group in Australia. The result was a failure ” group differences were small and clinically trivial”.
Report comment
I’ve been hearing voices for close to 20 years. Gave up on psychiatry & meds a decade ago. As mentioned two comments before this I also find MIA & psychiatry to be different sides of the same coin in too many areas. The same tired anti-psychiatry dogma carried forward for decades vs psychiatric arrogance and corruption along with Big Pharma & for profit insurance. Insurance companies now reporting huge profits assert their control over politics and medicine in equal measure. So far it doesn’t effect me.
Just finished “The Great Pretender“ by Susanna Cahalan about the Sanford psychologist “superstar” David Rosenhan. Consummate charmer, lecturer and con-artist whose 1970’s article on the infiltration of psych hospitals (by himself and “others“) that indirectly forced DSM changes (to avoid embarrassment), calling the public’s attention to conditions in and eventually the closing of many psych hospitals for better & much worse.
Cahalan was wrongly diagnosed Bipolar & then schizoaffective. An exceptional MD found the true cause as neurological, autoimmune encephalitis. She was one of the lucky ones. She was cured. Psychosis hasn’t reoccurred but she sees it as a constant threat.
She interviewed Stanford social psychologist, Lee Ross who had hallucinations from contracting Gillain-Barré syndrome. His eyelids were paralyzed, couldn’t close his eyes, was severely sleep deprived with the resulting auditory hallucinations. Ross’ quote in the book, “they like to say everybody’s about six degrees Fahrenheit from hallucinating.”
My point? The BBB is no longer impermeable. That’s a big deal (its possible even CV-19 gets through). The brain has its own immune system and links with the lower body which is now old science. So-called breakthroughs which aren’t, like psychosis as being categorized on a spectrum. Giving people the false impression of scientific medical neurological progress marching forever on-ward. I gave up on all of it and resort to making small changes, like complete 35 hour fasting once a week, cold showers, stopping coffee and alcohol (since April Fools Day, hah)
What is it about hearing voices and over consuming coffee? TCM claims just overdoing fluids makes you overly sentimental & emotional. I’ve heard hints that auditory hallucinations are said to be offset or eliminated by physical regimentation and planning every aspect of life. IOW, the opposite of the effects of hearing voices which tends to make me lazy. Sounds great but I refuse to turn my life into a tactical warrior boot camp.
Now it’s CV-19 in NYC. I was pretty much self isolating even before CV-19. Persistent voices, sudden heart jumping slamming doors, loud scrapping & base pounding auditory hallucinatory sounds etc even as I type. Street drugs, alcohol not wanted or needed. At best it’s called “spiritual emergency” (forget the spiritual part) at worst, lose of grey matter. You should see what it’s like just to compose this comment with iPad batteries cutting out. Even with that I have it relatively easy. The 7PM clapping and banging of pots and pans right now is happening in my apartment complex. Both real, upbeat and CV-19 first responder surreal.
Report comment
hi, i got diagnosed as schizophrenic, i experience auditory hallucinations off meds and what not. I hate being on antipsychotics cause i hate what they do to me, dumb me down, emotionally numb, lack of motivation etc. However i always get forced on meds cause parents call cops on me and then i get put into psychward and forced on meds. Recently ive been on a cto and was forced to take fluanxol for a year. however i luckilly got off that and have been med free for 3 to 4 months. So ive been on meds 4 seperate times b4, the first 3 times always 1-3 months. id be suffering from symptoms i listed above but would always recover from there about after a year. However this time im a little scared of if i will recover or not since ive been on meds for a year.
So im glad you posted this article, and i agree that better data would be shown if the patients tested were all newly diagnosed schizophrenics who have never been on meds b4, so that way we can accurately determine all the things the med is doing. for example is the medication causing cognitive decline, lack of emotion, and lack of motivation. I imagine all the ppl were already experiencing these symptoms as ive noticed all antipsychotics cause this. however maybe caplyta causes to lesser degree, we dont know since as the author pointed these clinical trials are mostly meaningless.
However i do plan on going on caplyta when i recover and go on psychosis again. As mentioned b4 i have no choice as i keep getting forced on meds. however i would rather try caplyta over a different medication since caplyta appears to antagonize the dopamine receptors to a lesser degree then current medications, also it binds to less receptors. I imagine i would still get side effects from this medication, but im hoping it will be less then previous medications i have tried. I wont know until i try the medication which prolly wont be for more then half a year.
on a side note, what do you think of the new antipsychotics coming out, ulotaront and karxt? im hoping it will be much better then current meds ive tried, as stated b4 i would rather live without taking antipsychotics, but when your schizophrenic and have psychosis, your basically forced to take antipsychotics. my only hope seems to be in better meds so i can live a normal life again.
Report comment
This post sheds light on the troubling trends surrounding new drug approvals for schizophrenia. It’s disheartening to see that many of these medications do not address patient needs effectively, often prioritizing profit over genuine innovation. I appreciate the critical examination of the pharmaceutical industry and hope it sparks more conversations about alternative approaches to treatment.
Report comment
This post really highlights the complexities and challenges surrounding new drug approvals for schizophrenia. It’s concerning to see how the focus often shifts from genuine patient needs to profit motives. We need more transparency and accountability in the approval process to ensure that treatments are both effective and beneficial for those struggling with this condition. Thank you for shedding light on such an important issue!
Report comment
This post sheds light on a critical issue in mental health care. The reliance on new drug approvals for schizophrenia often feels like a facade, lacking genuine innovation and effectiveness. It’s disheartening to see the system prioritize profit over patient well-being. We need more advocacy for holistic approaches that consider the complexities of mental health. Thank you for highlighting this important topic!
Report comment
This post sheds light on an essential issue that often gets overshadowed in mainstream discussions about schizophrenia treatment. The critique of how new drugs are approved raises critical questions about efficacy versus marketing. It’s disheartening to see patients caught in a cycle of ineffective treatments while the real needs for holistic care and innovative solutions are sidelined. Thank you for bringing attention to this vital conversation!
Report comment
This post sheds light on an important issue in mental health care. The way new drugs for schizophrenia are approved often feels more like a charade than a genuine effort to help patients. It’s crucial that we advocate for more transparency and effectiveness in these approvals, emphasizing patient well-being over pharmaceutical profits. Thank you for bringing this critical perspective to the forefront.
Report comment
This post raises critical points about the pharmaceutical industry and the often misleading narrative around new schizophrenia medications. It’s disheartening to see that many of these drugs are marketed with little evidence of significant benefit. We need more transparency in the approval process and better alternatives for those living with schizophrenia. Thank you for shedding light on this important issue!
Report comment
This post raises crucial points about the approval process for schizophrenia drugs. It’s alarming to see how often new medications are marketed with minimal evidence of real improvement in patient outcomes. We need to prioritize genuine advancements in treatment rather than merely cycling through new brand names. Thank you for shedding light on this important issue!
Report comment